«Количественный химический анализ вод. Методика выполнения измерений суммарных содержаний летучих фенолов в пробах природных и очищенных сточных вод экстракционно-фотометрическим методом после отгонки с паром. ПНД Ф 14.1:2.105-97»
Документ по состоянию на август 2014 г.
Утверждаю
Заместитель Председателя
Государственного комитета
Российской Федерации
по охране окружающей среды
А.А.СОЛОВЬЯНОВ
21 марта 1997 года
Методика допущена для целей государственного экологического контроля.
Методика рассмотрена и одобрена Главным управлением аналитического контроля и метрологического обеспечения природоохранной деятельности (ГУАК) и Главным метрологом Госкомэкологии России.
Настоящий документ устанавливает методику количественного химического анализа проб природных и очищенных сточных вод для определения в них массовой концентрации летучих фенолов в диапазоне от 2 до 30 мкг/куб. дм в пересчете на фенол фотометрическим методом после отгонки с водяным паром без разбавления и концентрирования пробы.
Если массовая концентрация летучих фенолов в анализируемой пробе превышает верхнюю границу, то допускается разбавление пробы таким образом, чтобы концентрация фенолов соответствовала регламентированному диапазону.
Мешающие влияния на определение фенолов могут оказать интенсивно окрашенные соединения кислого характера, а также сильные восстановители (например сульфиты), способные отгоняться с паром. Мешающее влияние других веществ устраняется в процессе отгонки.
Фотометрический метод определения массовой концентрации летучих фенолов основан на отгонке фенолов из подкисленной пробы воды, взаимодействии фенолов в отгоне с 4-аминоантипирином в присутствии гексацианоферрата (III) калия и экстракции образующегося окрашенного соединения хлороформом. Оптическую плотность экстракта измеряют на спектрофотометре (лямбда = 460 нм) или фотоэлектроколориметре (лямбда = 460 — 490 нм).
Согласно ГОСТ 27384 «Вода. Нормы погрешности измерений показателей состава и свойств» относительная погрешность измерений при доверительной вероятности Р = 0,95 не должна превышать значений, указанных в табл. 1.
Настоящая Методика количественного химического анализа обеспечивает получение результатов анализа с погрешностями, не превышающими значений, рассчитанных по соотношениям, приведенным в табл. 2.
При определении летучих фенолов в пробах с массовой концентрацией свыше 30 мкг/куб. дм после соответствующего разбавления погрешность измерения не превышает значений, рассчитанных по приведенным в таблице 2 зависимостям.
Метрологические характеристики приведены в виде зависимости от значения результата измерения массовой концентрации определяемого компонента в пробе — C.
Спектрофотометр или фотоколориметр, позволяющий измерять оптическую плотность при длине волны лямбда = 460 — 490 нм.
Кюветы с толщиной поглощающего слоя 50 мм.
Весы лабораторные 2 класса точности, ГОСТ 24104.
Весы технические лабораторные 4 класса точности, ГОСТ 24104, с пределом взвешивания 200 г.
СО с аттестованным содержанием фенола с погрешностью не более 1% при Р = 0,95 (или фенол, п. 4.2).
pH-метр типа pH-150, pH-155, ТУ 25-7410.003, ТУ 25-7416.0171 или иономер типа Анион-210, Анион-214.
Термометр с диапазоном 0 — 100 °C, ГОСТ 29224.
Шкаф сушильный общелабораторного назначения, ГОСТ 13474.
Плитки электрические с закрытой спиралью и регулируемой мощностью нагрева, ГОСТ 14919.
Колба для перегонки КП-1-50-19/26 ТХС, ГОСТ 25336.
Установка из стекла для перегонки растворителей (колба К-1-1000-29/32 ТС, дефлегматор 350-19/26-29-32 ТС, холодильник ХПТ-1-400-14/23 ХС), ГОСТ 25336.
Допускается использование других типов средств измерений, посуды и вспомогательного оборудования, в том числе импортных, с характеристиками не хуже, чем у приведенных в п. 4.1.
Фенол кристаллический, ГОСТ 6417.
Гидроксид натрия, ГОСТ 4328.
Сульфат меди, пентагидрат, ГОСТ 4165.
Аммиак водный, концентрированный, ГОСТ 3760.
4-Аминоантипирин, ТУ 6-09-3948.
Соляная кислота, ГОСТ 3118.
Тиосульфат натрия, пентагидрат, ГОСТ 27068.
Спирт этиловый, ГОСТ 18300.
Бумага индикаторная универсальная, ТУ 6-09-1181.
Фильтры обеззоленные «белая лента», ТУ 6-09-1678.
Вата хлопковая, ГОСТ 5556, или вата стеклянная.
Вода дистиллированная, ГОСТ 6709.
Все реактивы, используемые для анализа, должны быть квалификации ч.д.а. или х.ч.
Допускается использование реактивов, изготовленных по другой нормативно-технической документации, в том числе импортных, с квалификацией не ниже ч.д.а.
5.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.4.019.
5.2. Электробезопасность при работе с электроустановками по ГОСТ 12.1.019.
5.3. Организация обучения работающих безопасности труда по ГОСТ 12.0.004.
5.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой экстракционно-фотометрического анализа и изучивший инструкцию по эксплуатации спектрофотометра или фотоколориметра.
Измерения проводятся в нормальных лабораторных условиях.
Температура окружающего воздуха (22 +/- 6) °C.
Атмосферное давление (84 — 106) кПа.
Относительная влажность (80 +/- 5)%.
Частота переменного тока (50 +/- 1) Гц.
Напряжение в сети (220 +/- 10) В.
8.1. Посуду, предназначенную для отбора и хранения проб, промывают насыщенным раствором кальцинированной соды (карбоната натрия), а затем дистиллированной водой. При мытье сильно загрязненной посуды рекомендуется использовать хромовую смесь, после чего тщательно (не менее 20 раз) промывать водопроводной водой и споласкивать дистиллированной водой.
8.2. Пробы воды отбирают в стеклянные бутыли с плотно завинчивающимися пробками вместимостью 1 куб. дм.
Объем отбираемой пробы должен быть не менее 1 куб. дм.
8.3. Пробы анализируют не позже чем через 4 часа после отбора или в течение суток при условии хранения в холодильнике при t 10,2). На следующий день необходимо опять провести контроль pH и при необходимости довести его до нужной величины. Контроль следует осуществлять каждые 7 дней. Раствор устойчив при хранении в полиэтиленовой посуде.
9.1.2. Раствор 4-аминоантипирина, 2%.
1,0 г 4-аминоантипирина растворяют в 50 куб. см дистиллированной воды, фильтруют и переносят в посуду из темного стекла. Раствор хранят в холодильнике в течение 7 дней, при комнатной температуре в темном месте не более 3 дней. Для выполнения определений пригоден раствор, имеющий бледно-желтую окраску. При появлении темно-желтой или бурой окраски следует приготовить свежий раствор либо взять другой 4-аминоантипирин.
9.1.3. Раствор гексацианоферрата (III) калия, 8%.
Градуировочный раствор, аттестованный по процедуре приготовления, готовят из стандартного образца (СО) или кристаллического фенола.
При использовании СО производят разбавление исходного раствора в соответствии с инструкцией по его применению. Массовая концентрация фенола в градуировочном растворе должна составлять 5,00 мкг/куб. см. Хранят раствор в холодильнике не более 3 сут.
Приготовление градуировочного раствора из кристаллического фенола выполняют в соответствии с Приложением 2.
Для построения градуировочного графика необходимо приготовить образцы для градуировки с массовыми концентрациями фенола 0 — 30.0 мкг/куб. дм. Условия проведения анализа должны соответствовать п. 7.
Состав и количество образцов для градуировки для построения градуировочного графика приведены в табл. 3.
Для всех градуировочных растворов погрешности, обусловленные процедурой приготовления, не превышают 3% относительно приписанного значения массовой концентрации фенола.
При построении градуировочного графика в делительные воронки вместимостью 1000 куб. см помещают с помощью цилиндра 500 куб. см свежепрокипяченной и быстро охлажденной дистиллированной воды и приливают градуированной пипеткой вместимостью 5 куб. см аликвотные части градуировочного раствора фенола в соответствии с табл. 3.
Анализ образцов для градуировки проводят в порядке возрастания их концентрации согласно п. 10.
Измеряют оптическую плотность проб с добавками градуировочного раствора фенола и без него по отношению к хлороформу. Для построения градуированного графика каждую искусственную смесь необходимо фотометрировать 3 раза с целью исключения случайных результатов и усреднения данных. Усредненную оптическую плотность холостого опыта (проба, не содержащая фенола) вычитают из усредненной оптической плотности проб с добавками фенола.
Градуировочный график строят в координатах: массовая концентрация фенола, мкг/куб. дм — оптическая плотность.
Если условие стабильности градуировочной характеристики не выполняется только для одного образца для градуировки, необходимо выполнить повторное измерение этого образца с целью исключения результата, содержащего грубую погрешность.
Если градуировочная характеристика нестабильна, выясняют причины нестабильности градуировочной характеристики и повторяют контроль ее стабильности с использованием других образцов для градуировки, предусмотренных методикой. При повторном обнаружении нестабильности градуировочной характеристики строят новый градуировочный график.
Использованный хлороформ собирают в отдельную склянку и затем регенерируют. Для этого слив хлороформа помещают в делительную воронку вместимостью 1 куб. дм, добавляют равный объем дистиллированной воды и встряхивают воронку 2 мин. После расслоения фаз хлороформ переносят в другую воронку, вновь добавляют равный объем воды и повторяют промывание. После отстаивания хлороформ фильтруют через слой ваты или 2 — 3 неплотных бумажных фильтра в перегонную колбу. Перегоняют хлороформ, отбирая фракцию, кипящую при t = 60,5 — 62,0 °C. Первую порцию отгона, кипящую ниже 60,5 °C, возвращают в слив, а остаток после отгонки отбрасывают.
Мерным цилиндром отбирают 500 куб. см анализируемой воды и помещают ее в колбу для отгонки. Если в пробе присутствует активный хлор, приливают эквивалентное количество раствора тиосульфата натрия и дают постоять 5 мин. Добавляют 5 куб. см 10% раствора сульфата меди и 10 куб. см 10% раствора серной кислоты. Колбу помещают на электроплитку, присоединяют каплеуловитель и холодильник. Для уменьшения теплообмена колбу оборачивают асбестовым полотном или стеклотканью. Выходной отросток холодильника опускают в колбу вместимостью 500 куб. см, в которую предварительно помещают 10 куб. см раствора гидроксида натрия 0,05 моль/куб. дм. Нижний конец трубки холодильника должен быть погружен в этот раствор. При необходимости его можно удлинить, пристыковав вплотную к трубке холодильника стеклянную трубку нужной длины.
Нагрев колбы должен быть достаточно сильным так, чтобы отгонка пробы не превышала 3 ч, однако кипение пробы должно быть равномерным, спокойным; бурное кипение недопустимо. По мере увеличения объема отгона колбу опускают так, чтобы трубка холодильника не была погружена в отгон более чем на 3 см. Когда объем отгона в колбе составит около 460 куб. см (на колбе заранее следует сделать соответствующую метку), отгонку прекращают.
Отгон переносят в делительную воронку вместимостью 1000 куб. см, споласкивают колбу 30 — 40 куб. см дистиллированной воды и переносят ее в ту же воронку. Прибавляют 10 куб. см буферного раствора, 3 куб. см 2% раствора 4-аминоантипирина и 3 куб. см 8% раствора гексацианоферрата (III) калия, перемешивая пробу после добавления каждого раствора, и оставляют на 10 — 15 мин. Затем дважды экстрагируют пробу хлороформом, используя для первой экстракции 20 куб. см, второй — 10 куб. см хлороформа. Первую экстракцию выполняют в течение 2 мин., вторую — 1 мин. После расслоения фаз хлороформные экстракты фильтруют через комочек хлопковой или стеклянной ваты в мерную колбу или градуированную пробирку вместимостью 25 куб. см и доводят объем до метки хлороформом. Оптическую плотность экстракта измеряют на спектрофотометре при длине волны 460 нм или фотоэлектроколориметре при длине волны 460 — 490 нм в кюветах с толщиной поглощающего слоя 50 мм.
Одновременно с пробами выполняют холостой опыт, используя 500 куб. см свежепрокипяченной дистиллированной воды. Оптическую плотность холостого опыта вычитают из оптической плотности проб.
Если содержание фенолов превышает 30 мкг/куб. дм, для отгонки берут меньшую аликвоту анализируемой воды и разбавляют ее свежепрокипяченной дистиллированной водой до объема 500 куб. см.
Результат количественного анализа в документах, предусматривающих его использование, представляют в виде:
где ДЕЛЬТА — характеристика погрешности измерения для данной массовой концентрации фенолов (табл. 2).
Численные значения результата измерения должны оканчиваться цифрой того же разряда, что и значения характеристики погрешности.
Образцами для контроля являются реальные пробы природных и очищенных сточных вод. Объем отобранной для контроля пробы должен соответствовать удвоенному объему, необходимому для проведения анализа по методике. Отобранный объем делят на две равные части и анализируют в точном соответствии с прописью методики, максимально, варьируя условия проведения анализа, т.е. получают два результата анализа в разных лабораториях или в одной, используя при этом разные наборы мерной посуды, разные партии реактивов. Два результата анализа не должны отличаться друг от друга на величину допускаемых расхождений между результатами анализа:
При превышении норматива контрольное определение повторяют. При повторном превышении норматива выясняют причины, приводящие к неудовлетворительным результатам, и устраняют их.
Образцами для оперативного контроля точности являются стандартные образцы, близкие по составу анализируемым пробам.
ЗНАЧЕНИЯ НОРМАТИВОВ ОПЕРАТИВНОГО КОНТРОЛЯ ПОГРЕШНОСТИ ПРИ ПРОВЕДЕНИИ КОНТРОЛЯ С ИСПОЛЬЗОВАНИЕМ ОБРАЗЦОВ
Значение характеристик погрешности и нормативов контроля приведены в виде зависимости от значения результата измерения массовой концентрации определяемого компонента в пробе — C.
Приложение 1
(обязательное)
Приложение 2
(рекомендуемое)
1. Основной раствор фенола.
Навеску фенола около 0,1 г взвешивают на аналитических весах с точностью до четвертого знака после запятой, количественно переносят в мерную колбу вместимостью 50 куб. см с помощью этилового спирта, растворяют фенол, доводят раствор до метки и перемешивают. Массовую концентрацию фенола в полученном растворе рассчитывают по формуле:
C — массовая концентрация фенола в основном растворе, мг/куб. см;
Хранят раствор в склянке с плотно закрывающейся пробкой в холодильнике не более 6 месяцев.
Для приготовления раствора допускается использовать препарат бесцветный или со слаборозовой окраской, при более интенсивной окраске фенол следует перегонять. Установка для перегонки фенола изображена на рисунке (не приводится). Для уменьшения теплообмена колбу следует обернуть стеклотканью или асбестовым полотном. Для перегонки берут не более 1 г вещества. Первые две-три капли отгона отбрасывают, а следующую порцию собирают во взвешенный заранее на аналитических весах вместе с крышкой бюкс. После этого бюкс вновь взвешивают. Полученную навеску полностью используют для приготовления раствора. Контроль температуры не обязателен, однако для удобства можно вместо пробки закрыть колбу термометром со шлифом с соответствующим диапазоном температур. Температура кипения фенола 182 °C.
2. Промежуточный раствор фенола.
Рассчитывают объем основного раствора, который необходимо взять для получения раствора с массовой концентрацией фенола 100 мкг/куб. см:
V — объем основного раствора фенола, куб. см;
C — массовая концентрация фенола в основном растворе, мг/куб. см. Рассчитанный объем основного раствора градуированной пипеткой помещают в мерную колбу вместимостью 50 куб. см, доводят спиртом до метки и перемешивают. Хранят в холодильнике не более 1 мес.
3. Градуировочный раствор с массовой концентрацией фенола 5 мкг/куб. см.
Отбирают пипеткой с одной отметкой 5,0 куб. см промежуточного раствора фенола, переносят его в мерную колбу вместимостью 100 куб. см и доводят до метки дистиллированной водой. Срок хранения в холодильнике не более 3 сут.
источник
Настоящий документ устанавливает методику количественного химического анализа проб природных и очищенных сточных вод для определения в них массовой концентрации летучих фенолов в диапазоне от 2 до 25 мкг/дм 3 в пересчете на фенол экстракционно-фотометрическим методом без разбавления и концентрирования пробы.
Если массовая концентрация летучих фенолов в анализируемой пробе превышает верхнюю границу, то допускается разбавление пробы таким образом, чтобы концентрация фенолов соответствовала регламентированному диапазону.
Определению мешают интенсивно окрашенные соединения кислого характера, в частности нафтеновые кислоты при концентрации более 1 мг/дм 3 , гуминовые кислоты при концентрации более 2 мг/дм 3 , а также активный хлор.
Устранение мешающих влияний осуществляется в соответствии с п. 10.
Экстракционно-фотометрический метод определения массовой концентрации летучих фенолов основан на экстракции фенолов из воды бутилацетатом, реэкстракции их щелочью и образовании в реэкстракте окрашенных соединений фенолов с 4-аминоантипирином в присутствии гексацианоферрата (III) калия. Полученные соединения вновь экстрагируют бутил ацетатом и измеряют оптическую плотность экстракта на спектрофотометре (λ = 470 нм) или фотометре со светофильтром, имеющим максимум пропускания в диапазоне λ = 460 — 490 нм.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведённых в таблице 1.
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
Диапазон измерений, значения показателей точности, повторяемости, воспроизводимости, правильности
Диапазон измерений (в пересчете на фенол), мкг/дм 3
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ±δ, %
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемо сти), σ r %
Показатель воспроизводимости (относительное среднеквадрати ческое отклонение воспроизводи мости), σ R , %
Показатель правильности (границы относительной систематической погрешности при вероятности Р = 0,95), ±δс, %
Спектрофотометр или фотометр, позволяющий измерять оптическую плотность при длине волны λ = 460 — 490 нм
Кюветы с толщиной поглощающего слоя 50 мм
Весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г и ценой наименьшего деления 0,1 мг любого типа
Весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г и ценой наименьшего деления 10 мг любого типа
СО с аттестованным содержанием фенола с погрешностью не более 1 % при Р = 0,95 (или фенол, п. 4.3)
рН-метр или иономер с погрешностью измерения pH не более 0,05 единиц pH.
Термометр с диапазоном 0 — 100 °С
Плитки электрические с закрытой спиралью и регулируемой мощностью нагрева
Шкаф сушильный лабораторный с температурой нагрева до 130 °С
Стаканчики для взвешивания (бюксы)
Установка из стекла для перегонки растворителей в составе: колба К-1-1000-29/32 ТС, дефлегматор 350-19/26-29-32 ТС, холодильник ХПТ-1-400-14/23 ХС
Флаконы аптечные с навинчивающимися пробками и полиэтиленовыми вкладышами вместимостью 50 см 3
Средства измерений должны быть поверены в установленные сроки. Допускается использование других, в том числе импортных, средств измерений и вспомогательных устройств с характеристиками не хуже, чем у приведенных в п.п. 4.1 и 4.2.
Фенол кристаллический, очищенный
Бутиловый эфир уксусной кислоты (бутилацетат)
Карбонат натрия безводный Na2CО3,
или карбонат натрия декагидрат Na2CО3∙10H2О
Аммиак водный, концентрированный
Гексацианоферрат (III) калия K3[Fe(CN)6]
Сульфат натрия безводный Na2SО4
Тиосульфат натрия, пентагидрат
Бумага индикаторная универсальная
Фильтры обеззоленные «белая лента»
Вата хлопковая или вата стеклянная
Все реактивы, используемые для анализа, должны быть квалификации ч.д.а. или х.ч.
Допускается использование реактивов, изготовленных по другой нормативно-технической документации, в том числе импортных, с квалификацией не ниже ч.д.а.
5.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
5.2. Электробезопасность при работе с электроустановками обеспечивается по ГОСТ 12.1.019.
5.3. Организация обучения работающих безопасности труда проводится по ГОСТ 12.0.004.
5.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой экстракционно-фотометрического анализа и изучивший инструкцию по эксплуатации спектрофотометра или фотометра.
При выполнении измерений в лаборатории должны быть соблюдены следующие условия:
• температура окружающего воздуха
не более 80 % при температуре 25 °С;
8.1. Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб».
8.2. Посуду, предназначенную для отбора и хранения проб, промывают насыщенным раствором кальцинированной соды (карбоната натрия), а затем дистиллированной водой. При мытье сильно загрязненной посуды рекомендуется использовать хромовую смесь, после чего тщательно (не менее 20 раз) промывать водопроводной водой и споласкивать дистиллированной водой.
8.3. Пробы воды отбирают в стеклянные бутыли с плотно завинчивающимися пробками вместимостью 1 дм 3 .
Объем отбираемой пробы должен быть не менее 1 дм 3 .
8.4. Пробы анализируют не позднее, чем через 4 часа после отбора или в течение суток при условии хранения в холодильнике при t 3 с плотным полиэтиленовым вкладышем и завинчивающейся пробкой. Экстракт может храниться в темном прохладном месте в течение 1 месяца.
8.5. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— цель анализа, предполагаемые загрязнители,
— должность, фамилия отбирающего пробу, дата.
9.1. Приготовление растворов и реактивов
9.1.1. Аммонийно-аммиачный буферный раствор с pH 10,0 — 10,2.
50 г хлорида аммония растворяют в 50 см 3 дистиллированной воды, добавляют 350 см 3 концентрированного раствора аммиака и проверяют pH раствора рН-метром. Если значение pH раствора отличается от величины 10,0 — 10,2, необходимо добавить раствор аммиака (при pH 10,2). На следующий день необходимо опять провести контроль pH и при необходимости довести его до нужной величины. Контроль следует осуществлять каждые 7 дней. Раствор устойчив при хранении в полиэтиленовой посуде до 4 мес.
9.1.2. Раствор 4-аминоантипирина, 2 %.
1,0 г 4-аминоантипирина растворяют в 50 см 3 дистиллированной воды, фильтруют и переносят в посуду из темного стекла. Раствор хранят в холодильнике в течение 7 дней, при комнатной температуре в темном месте не более 3 дней. Для выполнения определений пригоден раствор, имеющий бледно-желтую окраску. При появлении темно-желтой или бурой окраски следует приготовить свежий раствор реактива, либо взять другой 4-аминоантипирин.
9.1.3. Раствор гексацианоферрата (III) калия, 8 %.
4 г K3[Fе(СN)6] растворяют в 50 см 3 дистиллированной воды, фильтруют, переносят в склянку из темного стекла. Раствор хранят в холодильнике в течение 7 дней, при комнатной температуре в темном месте не более 3 дней.
9.1.4. Раствор соляной кислоты, 1:1.
К 250 см 3 дистиллированной воды приливают 250 см 3 концентрированной соляной кислоты. Раствор устойчив при хранении в плотно закрытой посуде в течение 6 мес.
9.1.5. Раствор серной кислоты, 1:1.
К 100 см 3 дистиллированной воды, помещенной в термостойкий химический стакан, при непрерывном перемешивании приливают 100 см 3 концентрированной серной кислоты. Раствор охлаждают и переносят в толстостенную склянку. Раствор устойчив при хранении в плотно закрытой склянке в течение 1 года.
9.1.6. Раствор гидроксида натрия, 1 моль/дм 3 .
40 г NaOH растворяют в 1 дм 3 дистиллированной воды. Раствор устойчив при хранении в плотно закрытой полиэтиленовой посуде в течение 3 мес.
9.1.7. Раствор гидроксида натрия, 5 моль/дм 3 .
100 г NaOH растворяют в 500 см 3 дистиллированной воды. Раствор устойчив при хранении в плотно закрытой полиэтиленовой посуде в течение 3 мес.
9.1.8. Раствор карбоната натрия, 0,1 моль/дм 3 .
10,6 г Nа2СO3 или 28,6 г Na2CO3∙10H2O растворяют в 1 дм 3 дистиллированной воды. Раствор устойчив при хранении в плотно закрытой полиэтиленовой посуде в течение 6 мес.
9.1.9. Раствор тиосульфата натрия, 0,1 моль/дм 3 .
2,5 г Na2S2O3∙5H2O растворяют в 100 см 3 дистиллированной воды. Хранят в темной склянке не более 3 мес.
9.2. Приготовление градуировочного раствора
Градуировочный раствор, аттестованный по процедуре приготовления, готовят из стандартного образца (СО) или кристаллического фенола.
При использовании СО производят разбавление исходного раствора в соответствии с инструкцией по его применению. Массовая концентрация фенола в градуировочном растворе должна составлять 5,00 мкг/см 3 . Хранят раствор в холодильнике не более 3 суток.
Приготовление градуировочного раствора из кристаллического фенола выполняют в соответствии с Приложением Б.
9.3. Построение градуировочного графика
Для построения градуировочного графика необходимо приготовить образцы для градуировки с массовыми концентрациями фенола 0 — 25,0 мкг/дм 3 . Условия проведения анализа должны соответствовать п. 7.
Состав и количество образцов для градуировки для построения градуировочного графика приведены в табл. 2.
Для всех градуировочных растворов погрешности, обусловленные процедурой приготовления, не превышают 3 % относительно приписанного значения массовой концентрации фенола.
Состав и количество образцов для градуировки
при определении летучих фенолов
Концентрация фенола, мкг/дм 3
Объем градуировочного раствора, см 3
Объем дистиллированной воды, см 3
При построении градуировочного графика в делительные воронки вместимостью 1000 см 3 помещают с помощью мерного цилиндра 800 см 3 свежепрокипяченной и быстро охлажденной дистиллированной воды и приливают градуированными пипетками вместимостью 1 и 5 см 3 аликвотные части градуировочного раствора фенола в соответствии с табл. 2.
Анализ образцов для градуировки проводят в порядке возрастания их концентрации согласно п. 11.
Оптическую плотность проб с добавками градуировочного раствора фенола и без него измеряют по отношению к бутилацетату. Каждую пробу фотометрируют 3 раза с целью исключения случайных результатов и усреднения данных.
Усредненную оптическую плотность холостого опыта (проба, не содержащая фенола) вычитают из усредненной оптической плотности проб с добавками фенола.
Градуировочный график строят в координатах: массовая концентрация фенола, мкг/дм 3 , — оптическая плотность.
9.4. Контроль стабильности градуировочной характеристики
Контроль стабильности градуировочной характеристики проводят не реже одного раза в месяц или при смене основных реактивов (4-аминоантипирина, K3[Fе(СN)6], буферного раствора). Средствами контроля являются вновь приготовленные образцы для градуировки (не менее 3 образцов из приведенных в табл. 2).
Градуировочную характеристику считают стабильной при выполнении для каждого образца для градуировки следующего условия:
│Х — С│ ≤ 1,96 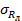 ,
,
где X — результат контрольного измерений массовой концентрации фенола в образце для градуировки;
С — аттестованное значение массовой концентрации фенола в образце для градуировки;
— среднеквадратическое отклонение внутрилабораторной прецизионности, установленное при реализации методики в лаборатории.
Примечание . Допустимо среднеквадратическое отклонение внутрилабораторной прецизионности при внедрении методики в лаборатории устанавливать на основе выражения: 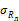 = 0,84σ R , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
= 0,84σ R , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Значения σ R приведены в табл. 1.
Если условие стабильности градуировочной характеристики не выполняется только для одного образца для градуировки, необходимо выполнить повторное измерение этого образца с целью исключения результата, содержащего грубую погрешность.
Если градуировочная характеристика нестабильна, выясняют причины нестабильности градуировочной характеристики и повторяют контроль ее стабильности с использованием других образцов для градуировки, предусмотренных методикой. При повторном обнаружении нестабильности градуировочной характеристики строят новый градуировочный график.
9.5. Регенерация бутилацетата
Использованный бутилацетат собирают в отдельную склянку и затем регенерируют. Для этого слив бутилацетата помещают в делительную воронку вместимостью 1 дм 3 , добавляют равный объем дистиллированной воды и встряхивают воронку 2 мин. После расслоения фаз воду из воронки удаляют, вновь добавляют равный объем воды и повторяют промывание. Вода после второго промывания должна иметь pH не выше 7. В противном случае промывание повторяют еще раз. После отстаивания воду как можно полнее удаляют, а бутилацетат фильтруют через слой ваты или 2 — 3 неплотных бумажных фильтра в перегонную колбу. Перегоняют бутилацетат, отбирая фракцию, кипящую при 108 °С. Первые 50 — 100 см 3 отгона возвращают в слив, а остаток после отгонки (около 50 см 3 ) отбрасывают.
Мешающее влияние нафтеновых и гуминовых кислот, окрашивающих бутилацетатный экстракт в бурый или коричневый цвет, устраняют промыванием экстракта перед реэкстракцией 50 см 3 раствора карбоната натрия 0,1 моль/дм 3 в течение 1 мин.
Если в пробе присутствует активный хлор, его удаляют, добавляя эквивалентное количество раствора тиосульфата натрия и давая постоять пробе 5 мин.
Мерным цилиндром вместимостью 1000 см 3 отбирают 800 см 3 анализируемой воды и помещают ее в делительную воронку вместимостью 1000 см 3 . Добавляют 40 г хлорида натрия, 1,5 см 3 раствора серной кислоты 1:1, 50 см 3 бутилацетата и экстрагируют фенолы в течение 3 мин. Дают пробе расслоиться в течение нескольких минут, сливают воду как можно полнее, затем круговыми движениями перемешивают экстракт и вновь удаляют отслоившуюся воду.
Экстракт переносят в делительную воронку вместимостью 250 см 3 , добавляют 50 см 3 раствора гидроксида натрия 1 моль/дм 3 и реэкстрагируют фенолы в течение 1 — 1,5 мин. После расслоения нижний водный слой переносят в делительную воронку вместимостью 100 см 3 , приливают 6 — 7 см 3 раствора соляной кислоты 1:1, затем добавляют эту же кислоту по каплям до pH 7 — 9 по универсальной индикаторной бумаге. Приливают 2 см 3 буферного раствора, затем по 1 см 3 растворов 4-аминоантипирина и гексацианоферрата (III) калия, перемешивая пробу после добавления каждого раствора.
Через 5 мин добавляют 23 см 3 бутилацетата и экстрагируют окрашенное соединение в течение 1 мин. После расслоения фаз нижний водный слой отбрасывают, а экстракт перемешивают круговыми движениями и дают отстояться еще 2 — 3 мин. Вновь удаляют водную фазу, а экстракт фильтруют через комочек хлопковой или стеклянной ваты в мерную колбу или пробирку вместимостью 25 см 3 . Объем экстракта доводят до метки бутилацетатом, одновременно промывая вату, через которую фильтровали экстракт. Аналогично выполняют холостой опыт, используя 800 см 3 свежепрокипяченной и охлажденной дистиллированной воды.
В тех случаях, когда при экстракции окрашенного соединения образуется очень стойкая, не расслаивающаяся в течение 15 — 20 мин эмульсия, экстракт из делительной воронки переносят в стакан вместимостью 50 см 3 , добавляют при непрерывном перемешивании палочкой безводный сульфат натрия до тех пор, пока не образуется прозрачный экстракт. Последний осторожно сливают в мерную колбу или пробирку, а сульфат натрия в стакане промывают 1 — 2 раза небольшим количеством бутилацетата, который переносят в ту же колбу или пробирку.
Оптическую плотность экстракта измеряют в кюветах с толщиной поглощающего слоя 50 мм на спектрофотометре при λ = 470 нм или на фотометре при λ = 460 — 490 нм относительно чистого растворителя. Оптическую плотность холостого опыта вычитают из оптической плотности проб.
Если массовая концентрация фенола в анализируемой воде превышает 25 мкг/дм 3 , пробу разбавляют свежепрокипяченной дистиллированной водой и анализируют повторно. В результат определения вводят соответствующую поправку.
При анализе сильно загрязненных и сильно эмульгирующихся вод пробу следует предварительно промыть хлороформом. Для этого к пробе в делительной воронке добавляют 16 см 3 раствора NaOH 5 моль/дм 3 , хлорид натрия, затем 50 см 3 хлороформа и экстрагируют 2 мин. После расслоения фаз нижний хлороформный слой сливают, добавляют к пробе 6 см 3 раствора серной кислоты 1:1, бутилацетат и далее проводят определение, как описано выше.
При таком промывании минерализованных проб на границе раздела фаз может образоваться объемный осадок гидроксидов, затрудняющий отделение хлороформа. В этом случае отделяют хлороформ до границы осадка, затем приливают. 4 см 3 раствора серной кислоты и осторожно перемешивают пробу, не переворачивая и не встряхивая воронку. Отслоившийся хлороформ быстро удаляют, после этого приливают остальное количество раствора серной кислоты
Массовую концентрацию летучих фенолов в анализируемой пробе воды X находят по градуировочному графику.
Если перед определением проводилось разбавление пробы, результат, найденный по градуировочному графику, умножают на коэффициент K = 800/V, где V — аликвота пробы воды, взятая для анализа, см 3 .
Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 3.
При превышении предела воспроизводимости могут быть использованы методы проверки приемлемости результатов анализа согласно раздела 5 ГОСТ Р ИСО 5725-6.
Значения предела воспроизводимости при вероятности Р = 0,95
Диапазон измерений (в пересчете на фенол), мкг/дм 3
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R , %
источник
Определение фенолов. При проведении анализа фенолы разделают на летучие с водяным паром (фенол, крезол, ксилинол) и нелетучие (ди- и триоксисоединения). Первая группа особенно важна по ее влиянию на вкус воды. Для определения фенолов этой группы проводят предварительную перегонку с водяным паром, а мешающие вещества, например H2S, удаляют осаждением.
Более старые аналитические методы определения фенолов были преимущественно колориметрическими. Но они не были одинаково применяемыми для определения всех фенола; для определения n-крезола и дихлорфенолов они не пригодны, поскольку применяемые реактивы не дают с этими фенолами достаточно интенсивно соединений. Так как получаемые окраски сравнивают с окраской, которую дает простой фенол, то в присутствии высших результаты определения не характеризуют содержание всех фенолов в пробе и их можно поэтому использовать только как ориентировочные.
Значительно более полную информацию о составе и содержании фенолов в пробе дают хромотографические методы разделения с последующим определением отдельных компонентов. Для этой цели применяются газовая, бумажная и хромотографии. Предложены автоматические приборы непрерывного измерения содержания фенолов по поглощению в УФ — области спектра. В качестве ускоренного метода может служить бромирование до m/n-бромфенолов с последующим измерением интенсивности образующегося помутнения. Бромирование, заканчивающееся нитрометрическим определением избытка брома, особенно пригодно для определения фенолов в высоких концентрациях. Заканчивать определение фенолов после бромирования можно также ИК — спектрометрией и газовой хромотографией.
Вследствие легкого окисления фенолов кислородом пробы по возможности необходимо анализировать сразу после их отбора.
Бромометрическое определение фенолов стандартным методом DVC. Определение общего содержания фенолов (более 100 мл). Экстрагируют 3 раза 500 мл нейтральной или слабокислой (рН=5) пробы воды бензолхинолиновой смесью (4:1 по объему), прибавляя каждый раз по 100 мл экстрагента. Отделенный слой органического растворителя взбалтывают 2 раза по 5 минут со 100 мл раствора NaOH. Щелочной экстракт осветляют кратковременным выпариванием, затем его встряхивают со 100 мл четыреххлористого углерода и органическую дозу отбрасывают. Объем полученного раствора фенолита доводят в мерной колбе вместимостью 500 мл водой до метки.
Аликвотную порцию 50 мл этого раствора переносят в колбу Эрленмейера, снабженную пришлифованной пробкой с присоединенной к ней капельной воронкой и через капельную воронку наливают 10 мл серной кислоты. После часового стояния добавляем через капельную воронку 10 мл 10% раствора йодида калия. Через 10 минут оттитровывают выделившийся йод 0,1 Н раствором Na2S2O3. Подобным же образом проводят холостое определение с 500 мл дистиллированной воды. Если разность между израсходованными объемами раствора совпадут в холостом опыте и при анализе проба превышает 10 мл, то 50 мл раствора фенолита перед бромированием разбавляют в 10 раз и из полученного раствора отбирают 50 мл для анализа. 1 мл 0,1 Н раствора Na2S2O3 соответствует 1,70 мг суммарного содержания фенолов (принимается, что средний молекулярный вес фенолов равен 102).
Реактивы. Раствор едкого натра, 3 1,15: раствор 160 г NaOH в 1 л воды. Серная кислота, S 1,12: добавляют 230 мл концентрированной H2SO4 к 1 л дистиллированной воды. Бромид-….. 0,1Н раствор: растворяют 2,784 высушенного KBrO3 rga и 10 г KBr в 1 л воды.
В работе в качестве экстрагенита применяют буцилацетат, который широко используется для извлечения фенолов из сточных вод газогенераторных станций. К 50 мл слабо подкисленной пробы сточной воды добавляют раствор CuSO4 и экстрагируют два раза буцилацетатом порциями по 40 мл. Экстракт буцилатетата взбалтывают три раза с порциями по 30 мл 10% раствора NaOН и титруют ….. часть полученного щелочного раствора фенолита бромид-…., как было описано выше.
Определение летучих с водяным паром фенолов (более 100 мг/л). Помещают 100 мл пробы сточной воды для перегонки с вертикальным холодильником и добавляют раствор CuSO4, чтобы связать H2S до тех пор, пока жидкость осадком не окрасится в синий цвет. Затем добавляют разбавленную CuSO4 (S 1,15) до растворения выпавшей в осадке синей гидроокиси меди. Перегоняют фенол в приемник, пока в перегонной колбе не останется 20 мл водной дозы; добавляют к последней 50 мл воды и снова перегоняют, пока не останется 20 мл. Дистиллят переливают в мерную колбу вместимостью 500 мл и разбавляют до метки. Отбирают оттуда 100 мл, бромируют, как описано ранее, 0,1Р раствором KBrO3 + KBr и титруют 0,1Н раствором Na2 S2O3.
Реактивы. Раствор сульфата меди: растворяют в воде 110 г CuSO4 5H2O и объем раствора доводят до 1 л. Серная кислота, S 1,15: добавляют 150 мл концентрированной H2SO4 и 1 л воды.
Определение колориметрическими методами. Определение с применением n-нитроанилина стандартным методом DEV.
Определение фенолов при содержании выше 0,1 мг/л. Помещают 200 мл анализируемой пробы в прибор для осадка фенолов, добавляют 1 мл раствора CuSO4, 1 мл CаSO4 (для связывания цианидов) и подкисляют добавлением 10 мл фосфорной кислоты (S 1,7). Полученный раствор перегоняют, собирая дистиллят в приемник, содержащий 10 мл 1 Н раствора Na2CO3. Перегонку ведут до тех пор, пока в перегонной колбе не останется 20 мл. Полученный дистиллят разбавляют до 250 мл.
В мерную колбу вместимостью 100 мл, содержащую 20 мл раствора n-нитроанилина, вносят несколько капель насыщенного раствора NaNO2 до полного обесцвечивания. Отобрав 50 мл дистиллята, смешивают с 30 мл 1Н раствора Na2CO3, выливают эту смесь в мерную колбу, содержащую раствор реагента, и доливают водой до 100 мл. Через 20 минут оптическую плотность раствора измеряют при = 530 нм по отношению к раствору, полученному в холостом определении. Калибровочную кривую строят по растворам фенола, содержащим от 0,1 до 10 мг/л.
Реактивы. Раствор сульфата меди: растворяют 10 г CuSO4 5H2O в 100 мл воды. Раствор сульфата кобальта: растворяют 10 г CаSO4Н2O в 100 мл воды. Раствор n-нитроанилина: растворяют 1,38 г n-нитроанилина в 310 мл 1 Н раствора HCl и доводят объем раствора до 2 л.
Экстракция полученного красителя бутанолом (для определения фенолов при содержании ниже 0,2 мг/л). Дистиллят, полученный перегонкой 200 мл пробы по описанному ранее способу (приемник содержит 30 мл 1Н раствора Na2CO3), переносят в делительную воронку вместимостью 500 мл и добавляют 20 мл раствора диазотированного n-нитроанилина. Через 20 минут образовавшийся … раствор встряхивают с 50 мл n -бутадиона и оптическую плотность бутанального раствора измеряют при = 530 нм. Калибровочную кривую строят по растворам, содержащим от 0,01 до 0,4 мг фенола в 1 л.
Определение с применением 4-аминоантипирина. Чувствительность определения достигает нескольких микрограммов в 1 л. Этот метод, как и другие фотометрические методы, не дают одинаково удовлетворительных результатов для всех галогенов фенола. Пара…. Фенолы этим способом не определяются.
Подготовка пробы. Для консервирования пробу воды подкисляют фосфорной кислотой (рН =4) и на каждый литр воды вносят 1 г CuSO4 5H2O для стерилизации и осаждения H2S. Окислители (например, свободный хлор) восстанавливают добавлением FeSO4.
Перегонка. Из 500 мл подкисленной пробы (рН =4) отгоняют 450 мл. Остаток в колбе разбавляют 50 мл воды и отгоняют еще 50 мл, затем оба отгона соединяют.
Прямое фотометрическое определение (содержание фенола 0,1- 5 мг/л). К 100 мл дистиллята добавляют 32 мл 5% раствора NH4, затем с помощью концентрированного раствора аммиака доводят рН раствора до 100,1, после чего добавляют 2 мл 2% свежеприготовленного водного раствора 4-аминоантипира и 2 мл 8% раствора K3Fe (CN)6. Через 15 минут измеряют оптическую плотность при = 510 нм по отношению к холостому раствору. Для построения комбинированных кривых используют растворы фенола, содержащие от 0 до 0,5 мг фенола в 100 мл.
Фотометрическое определение после экстракции (содержание фенола 0,005 — 0,1 мг/л).
- 1. К 500 мл дистиллята ( 25 мг/л) 100 мл пробы (рН=3-5) экстрагируют, встряхивая точно 10 мл изобутилового эфира уксусной кислоты, высушивают эфирный экстракт безводным Na2SO4 и 20 мкл полученного раствора впрыскивают в фрактометр 116 с термисторным детектором.
Условия определения. Колонка длиной 4 м, неподвижная фаза — 20% аниезан L на цианите, температура печи 180 °С, газ — носитель — гелий (55 мл/мин), относительное время удерживания (время удерживания фенола-10 мин — принято за 1): о-крезол — 1,53; м+n-крезол — 1,63; диметилфенолы — 2,2 -3,1; триметилфенолы — 3,8-4.
При определении фенолов в более низких концентрациях извлечение можно проводить диэтиловым эфиром. Эфирный экстракт упаривают до 0,5 мл.
Определение фенолов после галогирования. Повышение чувствительности газо-хромотографического определения фенолов можно достичь путем перевода фенолов в галогено-производные и анализа последних в хромотографе электронозахватным детектором.
Проще всего провести бромирование фенолов бромид-броматной смесью. Полученные бромфенолы извлекают изооктаном и разделяют их в хромотографе с электронозахватным детектором. Эти способом можно анализировать пробы, содержащие фенолы в концентрациях 5-15 мг/л при стандартном отклонении полученных результатов 1 мкг/л.
Содержащую фенолы слабокислую фракцию хлороформного экстракта обрабатывают б — бромпентафтортолуолом. Образующиеся пентафторбензиловые эфиры затем разделяют в хромотографе с электронозахватным детектором.
Было также предложено ацетилировать фенолы хлоруксусным ангидридом и полученные производные разделить в хромотографе с электронозахватным детектором.
Физико-химические методы определения ПАВ. Под детергентами или синтетическими поверхностно-активными веществами (С ПАВ) подразумевают активные моющие вещества, насыщающие поверхностное натяжение воды. Внешне их присутствие в воде обнаруживается по образованию более или менее устойчивой пены при движении воды или при ее взбалтывании. Наиболее важное применение этого класса соединений — в качестве моющих, чистящих и ополаскивающих средств. По своей химической структуре С ПАВ являются комбинацией водоотталкивающих (гидрофобных), но растворяющих жиры атомов углеводородов и растворимых в воде (гидрофильных) атомных групп. Различают анионоактивные, катионоактивные и неиногенные ПАВ. При анализе определяют отдельное содержание каждой из этих групп. В практическом отношении важнейшими среди них являются вещества, входящие в состав хозяйственных моющих порошков. Все они относятся к анионоактивной группе. Для некоторых специальных целей, например, в качестве дезинфицирующих средств, применяют иногда катионоактивные вещества. К неионогенным ПАВ относятся и некоторые природные продукты, например, саконины, которые раньше иногда применяли в качестве моющих средств. Синтетические неионогенные вещества имеют некоторые особенности, благодаря которым их стали все чаще применять в качестве моющих средств.
Разнообразное применение С ПАВ (для мытья тканей, посуды, очистки автомобилей, а также для личной гигиены) приводит к все увеличивающемуся переходу их в бытовые и производственные сточные воды. В отличие от обычного мыла они не осаждаются солями жесткости и поэтому в сточных водах их находят не в твердой фазе, а большей частью в растворе, причем чаще они стабилизируют суспензии, образованные различными нерастворимыми в воде загрязняющими веществами.
ПАВ в сточных и поверхностных водах оказывают разнообразное вредное воздействие. Образуемая ими пена достигает иногда метровой толщины и может препятствовать судоходству. Накапливаясь на поверхности воды, такая пена препятствует обмену веществ в водоеме, кроме того, как правило, пена оказывает токсическое действие на живые существа и, поэтому, уменьшает способность водоема к самоочищению.
Многие препараты С ПАВ, например, тетрапропиленбензолсульфанат, содержащие в своих молекулах сильно разветвленную цепочку углеводородов, очень устойчивы к химическому процессу окисления и поэтому они накапливаются в реках, нанося им большой ущерб. По закону 1962 г. все детергенты, которые биохимически окисляются меньше, чем на 80 («биологически жесткие») запрещены. Определение биохимической способности различных С ПАВ согласно узаконенным нормам стало важной задачей химиков, занимающихся анализом вод.
Определение анионоактивных веществ.
Определение с применением метиленового голубого. В делительной воронке вместимостью 250 мл в течение 1 минуты встряхивают 100 мл пробы воды, содержащей 20 — 250 мкг анионоактивных С ПАВ, с 10 мл фосфатного буферного раствора (рН=10), 5 мл нейтрального раствора метиленового голубого и 15 мл хлороформа. Отделившийся хлороформный слой взбалтывают во второй делительной воронке со смесью 100 мл дистиллированной воды и 5 мл кислого раствора метилового голубого и после расслоения фильтруют через пропитанную хлороформом вату в мерную колбу вместимостью 50 мл. Обработку в обеих делительных воронках повторяют 2 раза, встряхивая жидкость каждый раз с порциями хлороформа по 10 мл. Хлороформные вытяжки соединяют, доливают хлороформом до 50 мл, перемешивают и измеряют оптическую плотность при л = 650 нм на спектрофотометре или фотометре с интерференционным фильтром. Таким же способом проводят холостое определение.
Для вычисления результатов служит калибровочная кривая, построенная по растворам, содержащим 25, 50, 100, 150, 200 и 250 мкг тетрапропиленбензолсульфаната в 100 мл, что соответствует 10, 20, 40, 60, 80 и 100 мл эталонного раствора в 100 мл.
Реактивы. Нейтральный раствор метиленового голубого. Растворяют 0,35 г метиленового голубого в 1 л воды. Через 24 часа раствор пригоден к употреблению. Светопоглощение хлороформного раствора в холостом определении с этим реактивом не должно превышать 0,015 при толщине слоя 1 см.
Кислый раствор метиленового голубого. Растворяют 0,35 г метиленового голубого примерно в 500 мл воды, приливают 6,5 мл концентрированной серной кислоты и дополняют водой до 1 литра. Раствор готов к употреблению только через 24 часа. Светопоглощение хлороформного слоя в холостом опыте не должно превышать 0,015 при толщине слоя 1 см.
Фосфатный буферный раствор (рН=10). Растворяют 12,52 г двузамещенного кислого фосфата натрия Na2HPO4. 2H2O в 500 мл воды. Доводят значения рН раствора до 10 добавлением около 3 мл 0,5Н раствора NaOH и дополняют водой до 1 литра.
Эталонный раствор. Разбавляют 20 мл точно 5% раствора тетрапропиленбензолсульфаната (ТБС) водой до 1 л; отбирают 50 мл раствора и разбавляют до 1 л. Такой раствор содержит в 1 мл 2,5 мкг ТБС.
Если в пробе воды содержатся сульфиды, то сначала добавляют к ней щелочной буферный раствор, затем 2 мл 20% раствора перекиси водорода и оставляют на 5 минут.
Для удаления остатков ПАВ моющих средств стеклянные сосуды прополаскивают смесью 9 частей метанола или этанола и 1 части концентрированной HCl.
Лонгвелл и Манище при холостом определении этим способом нашли менее 0,01 мг/л, а при проведении анализа с добавлением известных синтетических моющих веществ к бытовым сточным водам определили их в количестве 87-97%.
Чтобы снизить значение холостого определения, рекомендуется нейтральный раствор метиленового голубого с буферным раствором непосредственно перед прибавлением к пробе воды и взболтать с хлороформом. Вместо фосфатного буферного раствора можно применять боротный, для приготовления которого смешивают равные объемы 0,05 мл раствора Na2B4O7 и 0,10 мл раствора NaCl. В результате этого воспроизводимость определений улучшается до 0,002 мг/л и из добавленного количества детергента (ТБС или диактилсульфасукцината) определяют уже 98-100%. Рекомендуется пользоваться хлороформом, не содержащем этиловый спирт.
Определение с применением других красителей.
Применение метилового зеленого. Практика показывает, что для определения ПАВ лучше применять метиловый зеленый, т.к. он меньше чувствителен к моющим веществам, чем метиловый голубой.
В делительную воронку наливают 20 мл пробы воды, добавляют 10 мл буферного раствора (рН=2,5),2 мл раствора метилового зеленого, точно 40 мл бензола и взбалтывают в течение 1 минуты. После этого дают жидкости расслоиться, спускают водный слой, промывают бензольный слой смесью 15 мл воды и 5 мл буферного раствора и оставляют на 20 — 30 минут до осветления. Не фильтруя, отбирают пипеткой бензольный раствор, переносят в кювету и измеряют оптическую плотность при л = 615 нм.
Реактивы. Раствор метилового зеленого. Растворяют 0,5 г красителя в 100 мл воды. Аббот рекомендует взбалтывать раствор реактива с хлороформом до тех пор, пока последний не будет окрашиваться.
Буферный раствор (рН-2,5). Растворяют 7,5 г NaCl в воде, разбавляют до 1 л и добавлением 0,1Н раствора HCl доводят рН раствора до 2,5.
Эталонный раствор. Раствор 5-100 мкг этого вещества в 20 мл должен при определении в кювете с толщиной слоя 1 см 4 при л = 615 нм показать оптическую плотность, равную 0,20. Стандартное отклонение 0,3 мкг в пробе, т.е. 0,015 мкг/л.
Применение азура А. Краситель азур А образует с ПАВ интенсивно окрашенные соединения и поэтому реактив особенно пригоден для определения незначительных количеств С ПАВ.
Энергично взбалтывают 50 мл пробы, содержащей 1-30 мкг анионо-активного вещества с 50 мл 0,1Н раствора H2SO4, 1 мл раствора азура А и точно 10 мл хлороформа в течение 2 минут. Хлороформный слой фильтруют через стеклянную вату в кювету и измеряют оптическую плотность при при л = 623 нм по отношению к хлороформу.
Реактивы. Раствор азура А. Растворяют 40 мг азура А в 5 мл 0,1Н раствора H2SO4 и разбавляют до 100 мл. Оптическая плотность хлороформного раствора, получаемого при обработке раствором азура А, 5 мкг ПАВ в 50 мл воды, равна 0,1, а при использовании метиленового голубого — только 0,03.
Определение стандартным методом ASTM D 2330. Этим методом можно пользоваться при определении только сульфатов, главным образом, алкиларилсульфонатов. Под действием таких сильных кислот, как HCl, гидролизующиеся детергенты, например, алкиловые эфиры серной кислоты, при кипячении разрушаются.
Пробу воды, содержащую не более 100 мкг ПАВ, кипятят с 30 мл концентрированной хлористоводородной кислоты около 1 часа и при этом почти досуха выпаривают. Затем разбавляют водой до 25 мл, вносят несколько капель фенофталеина, добавляют раствор едкого натра до щелочной реакции, кипятят 2 минуты, смывают в делительную воронку, разбавляют до 100 мл, добавляют серную кислоту до обесцвечивания индикатора, 10 мл буферного раствора (рН=7,5) и встряхивают 4 раза с порциями по 25 мл смеси, 100 мл хлороформа и 4 капель метилгентиламина. После этого выпаривают хлороформную вытяжку после добавления 5 мл 10% раствора NaOH, разбавляют до 100 мл водой и кипятят в течение 15 минут до полного улетучивания амина. Прибавляют фенолфталеин, нейтрализуют разбавленной H2SO4 и определяют содержание ПАВ описанным ранее методом Лонгвэлла и Маница с метиленовым голубым.
Фосфатный буферный раствор (рН = 7,5). Растворяют 10 г KH2PO4 в 800 мл воды, добавляют раствор NaOH до рН = 7,5 ± 0,1 и разбавляют до 1 литра.
Определение катионоактивных веществ стандартным методом DEV H 23.
Взбалтывают 100 мл пробы воды в делительной воронке с 10 мл цитратного буферного раствора, 5 мл 0,1Н раствора HCl, 2 мл бромфенолового синего и 50 мл хлороформа в течение 3 минут. Затем отделяют хлороформный слой, фильтруя его через маленький тампон из ваты. Первые 5 мл отбрасывают, оптическую плотность … измеряют при л = 416 нм. Калибровочную кривую строят по соответственно разбавленным эталонным растворам.
Метод пригоден для определения катионоактивных веществ в концентрациях, превышающих 0,05 мг/л.
Реактивы. Цитратный буферный раствор. Растворяют 21 г лимонной кислоты (С6Н8О7. Н2О) в 200 мл 1Н раствора NaOH и дополняют водой до 1 литра. Отбирают 309 мл этого раствора и доливают до 1 литра 0,1Н раствором HCl.
Раствор бромфенолового синего. Растворяют 0,150 г бромфенолового синего в 200 мл 0,01Н раствора NaOH и прибавляют 42 мл 0,1Н раствора HCl.
Стандартный раствор. Растворяют 1,00 г цетилтриметиламмония бромистого (или другого катионоактивного вещества, принятого за стандарт) в воде и разбавляют раствор до 1 литра. Отбирают 50 мл этого раствора и снова разбавляют до 1 литра; 50 мл полученного разбавленного раствора снова доводят до 1 литра. 1 мл такого эталонного раствора содержит 2,5 мнг бромистого цетилтриметиламмония.
Для определения истинного содержания катионоактивного вещества в используемом препарате 2 г последнего растворяют в воде и раствор доводят водой до 1 литра. К 100 мл полученного раствора прибавляют 25 мл 0,1Н раствора бихромата калия и через 2 часа отфильтровывают выпавший осадок через крупнопористый мембранный фильтр. В прозрачном фильтрате определяют избыток бихромата; для этого вносят в него 5 г йодида калия, 10 мл разбавленной (1:3) серной кислоты и через 10 минут титруют 0,1Н раствором Na2S2O3 в присутствии цинкиодкрахмального раствора.
Расчет. Содержание (q) катионоактивного вещества в процентах вычисляют по формуле:
где а — объем 0,1 Н раствора Na2S2O3, израсходованного на титрование в холостом опыте, мл;
b — то же, при титровании пробы, мл;
А — эквивалентный вес катионоактивного вещества (для бромистого цетилтриметиламмония — 36Н);
Определение неионогенных веществ.
Методы определения этой группы, как правило, основаны на предпосылке о необходимости предварительного удаления мешающих анионоактивных поверхностно-активных веществ. Эти вещества удаляют с помощью ионного обмена или же отделяют экстрагированием или пенообразованием неионогенных С ПАВ.
Для самого определения применяют ряд неорганических комплексообразователей, однако, часто вследствие взаимодействия их с веществами другого типа недостаточно специфичны.
Определения по Гею и Джинкинсу.
Неионогенные ПАВ выделяют из пробы воды путем пеноотделения. Через пробу воды продувают воздух; пузырьки воздуха проходят через бензольный слой и захватывают неионогенные ПАВ. Эти соединения обрабатывают роданокобальтатом, с которым они образуют комплексные соединения, последние разлагают и выделившийся роданид определяют фотометрическим методом Барка и Хигсана.
Применяемая для этих целей аппаратура состоит из стеклянной трубки (длиной 40 см) с 2 метками, ограничивающими объем 15 мл. Верхняя метка отстоит на 5 см от верхнего конца, который имеет желобок для слива. В нижний конец трубки вставлен стеклянный диск, через который можно барботировать воздух. В трубку наливают 100 мл пробы, подщелоченной 100 мл пробы, подщелоченной 10 каплями 1Н раствора NaOH. Воздух барбатируют со скоростью около 35-40 мл/мин. На водный слой наносят 15-20 мл бензола. После 3 часов барбатирования добавляют столько воды, чтобы бензольный слой оказался между метками, и доливают бензол, чтобы объем его составил 15 мл. Декантируют бензольный слой, не захватывая при этом вроду, в маленькую делительную воронку и взбалтывают 5 минут с 2 мл раствора родонокобальтата аммония. После расслоения декантируют 10 мл бензольного раствора, содержащего комплексное соединение (ПАВ-родонокобальтат), в центрифужную пробирку, центрифугируют, отбирают пипеткой аликватн7ую часть (7-8 мл) бензольного раствора в другую делительную воронку и смешивают с равным объемом воды, которая перед этим была подкислена 1 каплей концентрированной HCl. Встряхивают 5 минут, дают расслоиться и отбирают пипеткой 5 мл водной фазы, содержащей теперь только родонид, в мерную колбу вместимостью 10 мл и опредляют содержание родонида методом Барка и Хигсона. Калибровочную кривую строят по растворам, содержащим от 0 до 20 мл того ПАВ, которое предполагается присутствующим в пробе.
Родонокобальтат аммония. Растворяют 20 г NH4SCN, 3 г Co(NO3) 6H2O, 25 г NaCl в воде и доливают до 100 мл. Взбалтывают с бензолом и бензоловый слой отбрасывают. Если проба воды содержит также анионоактивные ПАВ, к 100 мл пробы воды в пеноудаляющей трубке вносят перед добавлением бензола 0,5 г подходящего анианита и продувают 5 минут воздух, при этом анианит связывает анионоактивные моющие вещества. Затем подщелачивают, вводят бензол и продолжают определение по методу Гея и Джинкинса.
Определение по Барку и Хигсану.
В мерную колбу вместимостью 10 мл к 5 мл пробы воды или к подкисленному хлористоводородной кислотой раствору после разложения анионоактивного вещества с помощью родонокобальтита добавляют каплю свежеприготовленной насыщенной бромной воды и хорошо перемешивают. Через 1-2 минуты удаляют избыток брома взбалтыванием с 0,2 мл 2% раствора As2O3, вносят 4 мл перемешанного раствора пиридина с дихлоридом n-фенилендиамина, дополняют водой до 10 мл, перемешивают, оставляют на 25 минут и измеряют оптическую плотность относительно чистой воды при л = 515 нм в кювете с толщиной слоя 1 см. Вычитают величину светопоглощения холостого определения с реактивами, которая не должна превышать Е = 0,05.
Раствор пиридина наливают 31 мл концентрированной HCl в смесь из 18 мл пиридина и 12 мл воды.
Раствор дихлорида и n-фенилендиамина. Растворяют 0,36 г твердого реактива в воде и раствор разбавляют до 100 мл.
Смешанный реактив. Смешивают 3 мл раствора пиридина и 1 мл раствор фенилендиамина.
Определение биохимически окисляющихся ПАВ.
Чтобы очистить реки от биохимически трудно окисляющихся моющих веществ, поступающих с производственными и бытовыми сточными водами, часто вызывающих образование пены высотой в метр и препятствующих процессу самоочищения в водоемах, в ФРГ было опубликовано постановление от 1 декабря 1962 г., согласно которому, начиная с 1 октября 1964 г. разрешалось производство только таких анионоактивных детергентов, которые биохимически окисляются по крайней мере на 80 %.
В лабораторной аппаратуре, моделирующей процесс очистки с помощью активного ила, смешивают питательный раствор с искусственно составленной сточной водой, содержащей 20 мг/л ПАВ в пересчете на активное вещество метиленового голубого (МГАВ) в соответствии с методом, описанным выше. При постоянном доступе воздуха, выводе образовавшегося активного ила и при непрерывной подаче свежего раствора, ежедневно определяют количество окислившихся органических веществ с метиленовым голубым.
Реактивы. Питательный раствор. В 1 л питьевой воды растворяют 3,75 г пентена из казеина, 2,50 г … экстракта, 0,650 г мочевины, 150 мг NaCl, 100 мг CaCl2. 2H2O и 50 мг MgSO4. 7Н2О.
Раствор детергента. В 1 л дистилированной воды растворяют столько исследуемого ПАВ (или моющего вещества), чтобы оно отвечало 9,60 г активного вещества метиленового голубого (МГАВ).
Искусственно приготовленная сточная вода. Разбавляют 1 л питательного раствора и 50 мл раствора детергента водопроводной водой до 24 литров. Содержание детергента в 1 л этого раствора равно 20 мг/л в пересчете на активное вещество метиленового голубого (МГАВ).
Посева бактерий не проводят, поскольку происходит естественная инфекция из окружающей среды.
Режим измерений. Приток воздуха регулируют таким образом, чтобы происходило глубокое перемешивание, но не выброс жидкости или выделение пены, и чтобы в сточной воде поддерживалось содержание растворенного кислорода, по крайней мере, 2 мг/л. Приток свежего и слив обработанного раствора устанавливается 1 л/ч.
Биологический ил из отстойника равномерно возвращается насосом в сосуд. После протекания процесса в течение 24 часов содержимое сборника хорошо перемешивают и отбирают пробу для определения содержания активного детергента. По полученному содержанию детергента, отнесенному к содержанию его свежей сточной воде, вычисляют среднее значение окисляемости. Для контроля окисления питательных веществ каждые два дня определяют перманганатную окисляемость. Органическая часть в сухом веществе биологического ила не должна превышать 3 г/л сточной воды; в противном случае соответствующий избыток удаляют.
источник