Настоящий нормативный документ устанавливает методику количественного химического анализа различных типов вод, с цепью измерения содержания взвешенных и прокаленных взвешенных веществ гравиметрическим методом. Методика распространяется на следующие объекты анализа: воды питьевые; воды природные, в том числе поверхностных и подземных источников водоснабжения; воды сточные производственные, хозяйственно-бытовые, ливневые и очищенные. Методика может быть использована д ля анализа проб снежного покрова и талых вод.
Диапазон измерений содержания взвешенных и прокаленных взвешенных веществ от 0,5 до 5000 мг/дм 3 .
Продолжительность анализа одной пробы на содержание взвешенных веществ 14 часов, серии из 10 образцов — 15 часов.
Продолжительность анализа одной пробы на содержание прокаленных взвешенных веществ 17 часов, серии из 10 образцов — 18 часов.
Блок-схема проведения анализа приведена в приложении.
Определению мешают значительные количества масел и жиров, поэтому при отборе пробы должно быть исключено попадание в нее поверхностной пленки или кусочков жира. Если все-таки в пробе, доставленной в лабораторию, на поверхности присутствуют видимые жир или масло, то перед проведением анализа их удаляют. Жир с поверхности отобранной пробы снимают ложкой или шпателем, а масло кусочком фильтровальной бумаги.
Удаляют так же загрязнения в виде единичных включений, например, мелкие палочки, траву и т.п.
Содержание прокаленных взвешенных веществ дает ориентировочное представление о минеральном составе взвеси в воде, а потери при прокаливании, т.е. разность между массой взвешенных и прокаленных взвешенных веществ — о количестве органических соединений во взвеси.
ГОСТ 12.0.004-90 Система стандартов безопасности труда. Организация обучения безопасности труда. Общие положения
ГОСТ 12.1.004-91 Система стандартов безопасности труда. Пожарная безопасность. Общие требования
ГОСТ 12.1.007-76 Система стандартов безопасности труда. Вредные вещества. Классификация и общие требования безопасности
ГОСТ 12.4.009-83 Система стандартов безопасности труда. Пожарная техника для защиты объектов. Основные виды. Размещение и обслуживание
ГОСТ 17.1.5.05-85 Охрана природы. Гидросфера. Общие требования к отбору проб поверхностных и морских вод, льда и атмосферных осадков
ГОСТ 1770-74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Общие технические условия
ГОСТ 3118-77 Реактивы. Кислота соляная. Технические условия
ГОСТ 4147-74 Реактивы. Железо (III) хлорид 6-водный. Технические условия
ГОСТ 6709-72 Вода дистиллированная. Технические условия
ГОСТ 9147-80 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 25336-82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 27384-2002 Вода. Нормы погрешностей измерений показателей состава и свойств
ГОСТ Р 12.1.019-2009. Система стандартов безопасности труда. Электробезопасность. Общие требования и номенклатура видов защиты
ГОСТ Р ИСО 5725-6-2002. Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности
ГОСТ Р 50779.42-99 (ИСО 8258-91) Статистические методы. Контрольные карты Шухарта
ГОСТ Р 51592-2000 Вода. Общие требования к отбору проб
ГОСТ Р 52501-2005 Вода для лабораторного анализа. Технические условия
ГОСТ Р 53228-2008 Весы неавтоматического действия. Часть 1. Метрологические и технические требования. Испытания
Настоящая методика обеспечивает получение результатов измерений с погрешностью, не превышающей значений, приведенных в таблице 1, при доверительной вероятности 0,95. Приписанные погрешности измерений не превышают нормы погрешностей, установленные ГОСТ 27384.
Методика определения взвешенных веществ основана на выделении их из пробы путем фильтрования воды через предварительно взвешенный бумажный или мембранный фильтр и определении веса осадка на фильтре, высушенного до постоянной массы при (105 ± 2) °С.
Методика определения прокаленных взвешенных веществ основана на выделении их из пробы путем фильтрования воды через предварительно взвешенный бумажный или мембранный фильтр, высушивании до постоянной массы при (105 ± 2) °С и далее определении веса осадка на фильтре, прокаленного до постоянной массы в муфельной печи при (600 ± 15) °С.
Таблица 1 — Значения показателей повторяемости, воспроизводимости и точности
Диапазон измерений, мг/дм 3
Показатель повторяемости (стандартное отклонение повторяемости), σ r , %
Показатель воспроизводимости (стандартное отклонение воспроизводимости) σ R , %
Показатель точности (границы относительной погрешности при Р = 0,95), ±δ, %
Прокаленные взвешенные вещества
Примечание — Показатель точности измерений соответствует расширенной неопределенности при коэффициенте охвата k = 2.
5.1.1 Бюксы стеклянные СН-45/13, СН-60/14 по ГОСТ 25336.
5.1.2 Весы лабораторные с максимальной нагрузкой 210 г высокого класса точности по ГОСТ Р 53228.
5.1.3 Воронки лабораторные, В-56-80 ХС, В-75-110 ХС по ГОСТ 25336.
5.1.4 Гомогенизатор, например, марки IKA фирмы Labortechnic (Германия), модель Ultra-Turrax Т 25 или любой другой.
5.1.5 Дистиллятор или установка любого типа для получения воды дистиллированной по ГОСТ 6709 или воды для лабораторного анализа степени чистоты 2 по ГОСТ Р 52501.
5.1.6 Колбы конические вместимостью 500 и 1000 см 3 по ГОСТ 25336.
5.1.7 Печь муфельная с рабочей камерой футерованной керамическим муфелем, обеспечивающая температуру (600 ± 15) °С.
5.1.8 Пинцет металлический с острыми концами.
5.1.10 Установка фильтровальная с вакуумным насосом.
5.1.11 Флаконы с притертой пробкой (для хранения растворов реактивов).
5.1.12 Холодильник бытовой, обеспечивающий хранение проб при температуре (2 — 10) °С.
5.1.13 Цилиндры мерные вместимостью 500 и 1000 см 3 по ГОСТ 1770, 2 класса точности.
5.1.14 Шкаф сушильный общелабораторного назначения, обеспечивающий температуру (105 ± 2) °С.
5.1.16 Шпатель или ложка любая.
Допускается использование средств измерения, вспомогательного оборудования, лабораторной посуды с аналогичными или лучшими метрологическими и техническими характеристиками.
5.2.1 Вода дистиллированная по ГОСТ 6709 или для лабораторного анализа по ГОСТ Р 52501 (2-ой степени чистоты), (далее вода дистиллированная).
5.2.2 Кислота соляная по ГОСТ 3118, х.ч.
5.2.3 Железо (III) хлорид (хлорное железо), 6-водное по ГОСТ 4147, ч., насыщенный раствор (для маркировки бюксов).
5.2.5 Фильтры мембранные с диаметром пор 0,45 мкм.
Допускается использование реактивов более высокой квалификации, а также материалов с аналогичными или лучшими характеристиками.
6.1 При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
6.2 При работе с оборудованием необходимо соблюдать требования электробезопасности при работе с электроустановками по ГОСТ Р 12.1.019 и требования техники безопасности при работе с муфельной печью в соответствии с инструкцией по эксплуатации.
6.3 Организация обучения работающих безопасности труда должна проводиться по ГОСТ 12.0.004.
6.4 Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
К выполнению измерений и обработке их результатов допускают лиц, владеющих техникой гравиметрического анализа.
При выполнении измерений в лаборатории должны быть соблюдены следующие условия:
относительная влажность воздуха
9.1 Отбор проб производится в соответствии с ГОСТ Р 51592 и ГОСТ Р 51593. Отбор проб воды осуществляют в стеклянные или пластиковые флаконы. Пробы снега в соответствии с ГОСТ 17.1.5.05 переводят в талую воду при комнатной температуре.
9.2 Объём отбираемый пробы должен быть (1000 — 2000) см 3 для питьевой и природной воды и не менее 250 см 3 — для сточной или загрязненной пробы воды.
9.3 Срок хранения пробы 24 часа при температуре (2 — 10) °С.
9.4 При отборе проб составляют сопроводительный документ по утвержденной форме, в котором указывают:
— место, дату и время отбора;
— должность, фамилию сотрудника, отбирающего пробу.
30 см 3 соляной кислоты смешивают со 170 см 3 дистиллированной воды. Смесь хранят под тягой во флаконе с притертой пробкой. Срок хранения — 6 месяцев.
Тонкой деревянной палочкой или спичкой на фарфоровые тигли наносят идентификационные метки (номера) насыщенным раствором хлорного железа. Затем тигли ставят в муфельную печь, предварительно нагретую до (600 ± 15) °С на (5 — 10) минут. Метки приобретают коричневую окраску и не смываются водой и растворами кислот.
10.2.2 Прокаливание и взвешивание тиглей
Промаркированные фарфоровые тигли промывают раствором соляной кислоты, приготовленной по 10.1, затем — дистиллированной водой, подсушивают на воздухе и прокаливают при (600 ± 15) °С в течение 1 часа, охлаждают в эксикаторе и взвешивают. Прокаливание повторяют до тех пор, пока разница между двумя последними взвешиваниями не будет превышать 0,5 мг. Значение массы тигля записывают в рабочем журнале.
Примечание — Если одни и те же тигли используют ежедневно, при этом их массы изменяются в допустимых пределах (± 0,5 мг) достаточно проведения одного прокаливания в течение часа.
В маркированные стеклянные бюксы помещают мембранные фильтры и сушат в сушильном шкафу при температуре (105 ± 2) °С не менее 1 часа. Допускается выдерживание фильтров в сушильном шкафу в течение (3 — 5) часов. Значение массы бюкса с фильтром записывают в рабочем журнале. Если фильтры не были использованы в течение рабочего дня, процедуру сушки повторяют на следующий день, причем первая выдержка в сушильном шкафу должна быть не менее 1 часа.
Примечание — При необходимости подготавливают к проведению анализа мембранные фильтры в соответствии с инструкцией по их применению.
Фильтры складывают конусом по форме воронки, и промывают на воронке 250 — 300 см 3 дистиллированной воды. Фильтры подсушивают на воздухе досуха, вынимают из воронки, помещают в сложенном виде в маркированный бюкс и сушат в сушильном шкафу при (105 ± 2) °С с открытой крышкой не менее 2,5 часов.
Примечание — Если промытый фильтр после высушивания на воздухе остается слегка влажным, то на его высушивание до постоянной массы в сушильном шкафу потребуется не менее 5 часов.
Затем закрывают бюкс крышкой, охлаждают в эксикаторе и взвешивают на аналитических весах. Повторяют процедуру сушки, с выдержками в сушильном шкафу по 30 минут до тех пор, пока разница между двумя последними результатами взвешивания не будет превышать 0,5 мг. Значение массы бюкса с фильтром записывают в рабочем журнале после каждого взвешивания. Последний результат взвешивания используют для расчетов.
Подготовленные к анализу фильтры хранят в закрытом эксикаторе не более 7 дней. В течение указанного срока хранения повторное взвешивание фильтра перед фильтрованием не требуется. По окончании срока хранения фильтры высушивают ещё раз при (105 ± 2) °С в течение 1 часа. Значение массы бюкса с фильтром записывают в рабочем журнале.
Перед проведением анализа пробу тщательно гомогенизируют. В зависимости от ожидаемого содержания взвешенных веществ для анализа используют от 50 до 2000 см 3 пробы. Объём пробы подбирают таким образом, чтобы масса взвешенных веществ на фильтре (привеса) составляла не менее 0,0010 г. Питьевую и природную воду рекомендуется фильтровать через мембранный, а сточную воду через бумажный фильтр.
Через подготовленный фильтр пропускают анализируемую воду.
При использовании мембранного фильтра частицы, приставшие к стенке воронки для фильтрования, смывают на фильтр дважды порциями фильтрата по 10 см 3 .
При работе с бумажным фильтром фильтр с осадком трижды промывают дистиллированной водой порциями по 10 см 3 .
Фильтр подсушивают на воздухе (2 — 3) часа и помещают в тот же бюкс, где проводилось предварительное взвешивание. Мембранный фильтр высушивают в течение 1 часа, а бумажный в течение 4 часов в сушильном шкафу при (105 ± 2) °С. Фильтр с бюксом охлаждают в эксикаторе и взвешивают. Повторяют процедуру сушки до тех пор, пока разница между двумя последними результатами взвешиваниями не будет превышать 0,5 мг. Значения каждого взвешивания записывают в рабочий журнал.
Высушенный фильтр (11.1) с взвешенными веществами помещают в подготовленный фарфоровый тигель, который затем устанавливают в муфельную печь и прокаливают при температуре (600 ± 15) °С в течение 1 часа. Тигель охлаждают в эксикаторе и взвешивают. Повторные прокаливания проводят в течение 1 часа до тех пор, пока разница между результатами двух последних взвешиваний не будет превышать 0,5 мг. Значения каждого взвешивания записывают в рабочий журнал.
Примечание — В случае, если требуется определение только прокаленных веществ, то доведение бюкса с фильтром после высушивания при (105 ± 2) °С до постоянной массы не требуется. После высушивания фильтра в сушильном в шкафу, его сразу помещают в подготовленный тигель и далее проводят анализ как описано в 11.2.
12.1 Содержание взвешенных веществ в анализируемой пробе воды рассчитывают по формуле
где X1 — содержание взвешенных веществ, мг/дм 3 ;
m2 — масса бюкса с мембранным или бумажным фильтром со взвешенными веществами, г;
m1 — масса бюкса с подготовленным мембранным или бумажным фильтром, г;
V — объём пробы воды, взятой для анализа, дм 3 .
12.2 Содержание прокаленных взвешенных веществ в анализируемой пробе воды рассчитывают по формуле
где Х2 — содержание прокаленных взвешенных веществ, мг/дм 3 ;
m4 — масса тигля с остатком после прокаливания, г;
m3 — масса прокаленного тигля, г;
m — масса золы бумажного фильтра (указана на упаковке фильтра), г;
Примечание — В случае использования мембранного фильтра масса золы не учитывается.
V — объём пробы воды, взятой для анализа, дм 3 .
Результаты количественного анализа в протоколах анализов представляют в виде:
где Δ = δ × 0,01 × X — значение характеристики погрешности (см. таблицу 1).
Результат анализа округляют с точностью: при содержании взвешенных и/или прокаленных взвешенных веществ
14.1 При получении двух результатов измерений (X1, Х2) в условиях повторяемости (сходимости) осуществляют проверку приемлемости результатов в соответствии с требованиями ГОСТ Р ИСО 5725-6 (раздел 5).
Результат измерений считают приемлемым при выполнении условия:
Значения пределов повторяемости (r) приведены в таблице 2.
14.2 При получении результатов измерений в двух лабораториях (Хлаб1, Хлаб2) проводят проверку приемлемости результатов измерений в соответствии с требованиями ГОСТ Р ИСО 5725-6 (раздел 5).
Результат измерений считают приемлемым при выполнении условия:
Значения пределов воспроизводимости (R) приведены в таблице 2.
Контроль точности результатов измерений при реализации методики в лаборатории проводят с использованием рабочих проб.
При регулярном выполнении анализов по методике проводят контроль стабильности среднеквадратического отклонения внутрилабораторной прецизионности с использованием контрольных карт с периодичностью, установленной в лаборатории. Расчет контрольных границ проводят в соответствии с рекомендациями ГОСТ Р 50779.42 и ГОСТ Р ИСО 5725.
При эпизодическом выполнении анализов по методике проводят оперативный контроль показателя повторяемости. Для этого одну пробу из серии рабочих проб тщательно гомогенизируют, делят на две части и проводят анализ в условиях повторяемости. Далее результаты оценивают по 14.1.
Таблица 2 — Пределы повторяемости и воспроизводимости результатов измерений (при вероятности Р = 0,95)
Диапазон измерений, мг/дм 3
Предел повторяемости (при n = 2 и Р = 0,95), r ,%
Предел воспроизводимости (при n = 2 и Р = 0,95), R , %
источник
46.Методы анализа воды: гравиметрические, титриметрические, фотометрические, потенциометрические, вольтамперометрические.
Гравиметрический – основан на определении массы вещества. В ходе анализа вещество отгоняется в виде какого-либо летучего соединения или осаждается из раствора в виде малорастворимого соединения. Осадок взвешивается в виде соединения строго определенного состава, весовая форма по составу совпадает с осаждаемой. По весу высушенного или прокаленного осадка вычисляется содержание определенного компонента в данном образце. Достоинства: высокая точность, отсутствие необходимости калибровки, простота. Недостатки: значительный расход времени на выполнение анализа.
Титриметрический. Основан на точном измерении количества реактива израсходованного на реакцию с определенными веществами. Титрированный раствор – раствор, концентрация которого известна с высокой точностью. Титрование – прибавление титрованного раствора к анализируемому для точного определения эквивалентного количества. Момент титрирования – точка эквивалентности. Титрирующий раствор – титрант. Используются реакции кислотно-основного взаимодействия, удовлетворяющие требованиям, которые предъявляются к титриметрическим реакциям. Взаимодействие должно происходить полностью и с высокой скоростью. Достоинства: быстрота выполнения, простота оборудования, удобство выполнения серийных анализов, большой набор химических реакций. Недостатки: необходимость предварительной стандартофикации растворов титранта и калибровки мерной посуды.
Фотометрический. Измеряет поглощение света анализируемым раствором обычно после введения в него реактива, реагирующего с определенным компонентом сточной воды с образованием интенсивно поглощающего свет соединения. Приборы: Источник света – светофильтр – кювета с раствором – детектор. Конструкция прибора зависит от области спектра применения. Излучение выбирают такое, что бы соединение имело max светопоглощение, а примеси – min. Достоинства – широкая область применения, высокая чувствительность. Недостатки: калибровка аппаратуры, посуды.
Потенциометрия и потенциометрическое титрование. Потенциометрия основана на измерении небольших равновесных напряжений между электродами гальванической ячейки. Метод можно применять для установления активности веществ в растворе (прямая потенциометрия) и для нахождения точки эквивалентности при титриметрических определениях (потенциометрическое титрование). Прямая ПМ находит применение при определении рН растворов, а также многих ионов с использованием ионоселективных электродов. В анализе природных вод и питьевой воды ионоселективные электроды применяют для определения кадмия, меди, свинца, серебра, щелочных металлов, бромид-, хлорид-, цианид-, фторид-, иодид — и сульфид-ионов.
Вольтамперометрические методы анализа. Это совокупность методов исследования кривых ток-потенциал и их зависимостей от электродных реакций и концентраций определяемых веществ. Один из основных ВАМ методов – полярография. Метод заключается в получении и анализе кривых ток-потенциал на ртутном капельном электроде. Методом полярографии можно определить любые вещества, способные к эл-хим превращениям на электродах. Качественная информация следует из значения потенциала полуволны (φ1/2), количественная – из определения высоты волны (id).
Типичная полярографическая волна, используемая для качественного и количественного определения электродно-активных веществ.
источник
Гравиметрический метод анализа является одним из важнейших методов количественного химического анализа. Название «гравиметрический» происходит от латинского слова «gravitas» (вес), поэтому метод называют также весовым анализом.
Основоположником гравиметрического анализа является шведский ученый У.Т. Бергман. В 1780 г. он опубликовал четыре книги, в которых были впервые систематизированы основы качественного и количественного анализа и заложены основы гравиметрического анализа в растворах.
В истории химии гравиметрический анализ (г.а.) сыграл выдающуюся роль. С его помощью были установлены все основные химические законы и состав химических соединений.
Основным достоинством г.а., как уже отмечалось в разделе 2.1, является его высокая точность (до 0,2 %). Это намного превышает точность титриметрических (см. раздел 2.1) и инструментальных (см. раздел 8) методов анализа. Однако, превосходя эти методы по точности, г.а. сильно уступает им по длительности процедур анализа. Из-за большой продолжительности гравиметрические определения не могут использоваться для экспрессного (быстрого) определения каких-либо показателей качества сырья или готовой продукции, а также параметров технологического процесса ее получения. В то же время г.а. незаменим при арбитражных (спорных) анализах и широко применяется при выполнении научно-исследовательских работ для сравнения аналитических данных, полученных разными методами.
В технохимическом контроле в пищевой промышленности гравиметрические определения находят такое же ограниченное применение, как и во всей современной аналитической практике. В основном с помощью г.а. устанавливают содержание влаги и золы в пищевых продуктах, а также в сырье. Эти показатели называются соответственно «массовая доля влаги» и «массовая доля золы (зольность)» и являются одними из основных физико-химических показателей, определяющих качество сырья и готовых продуктов питания. Например, по количеству золы, получаемой при сжигании муки, муку стандартизуют по сортам. Так, массовая доля золы пшеничной муки высшего сорта равна 0,55 %, I сорта — 0,75 %, II сорта — 1,25 %, пшеничной обойной — 1,9 %. Еще одной областью применения г.а. в аналитической химии пищевых продуктов можно считать апробирование и установление критериев точности новых методов и новых методик анализа сырья и готовой продукции пищевой промышленности.
3.1. Сущность гравиметрического анализа
Напомним (см. раздел 2.1), что гравиметрический анализ — это метод количественного химического анализа, основанный на точном измерении массы определяемого вещества или его составных частей, выделяемых в химически чистом состоянии или в виде соответствующих соединений точно известного постоянного состава.
Таким образом, при гравиметрическом анализе из навески вещества или образца получают осадок или остаток, который взвешивают.
Метрологические характеристики г.а. приведены в разделе 2.1.
Предел обнаружения г.а. ограничивается растворимостью осадка и чувствительностью аналитических весов.
3.2. Классификация методов гравиметрического анализа
Все многочисленные гравиметрические определения можно разделить на три большие группы:
В методах выделения определяемый компонент выделяют в свободном состоянии из анализируемого образца и взвешивают на аналитических весах. Так, например, определяют массовую долю золы в пищевых продуктах (в частности, уже упоминавшуюся выше зольность муки) или массовую долю сухих веществ (сухого остатка).
В методах отгонки г.а. определяемый компонент количественно отгоняют в виде летучего соединения путем нагревания анализируемого образца или действием соответствующих реагентов.
Методы отгонки бывают прямыми и косвенными.
В прямых методах отгонки определяемый летучий компонент поглощают специфическим поглотителем и по увеличению его массы вычисляют содержание определяемого компонента, или определяемое вещество отгоняют из смеси и образовавшийся отгон взвешивают.
В косвенных методах отгонки определяемое вещество отгоняют из точной навески анализируемого образца. После окончания отгонки образец снова взвешивают. Массу определяемого вещества находят по разности масс образца до и после отгона О.В. В пищевой промышленности таким образцом определяют массовую долю влаги в различных видах сырья и готовых изделиях. Например, в хлебопекарной промышленности анализируют на этот показатель муку, практически все дополнительное сырье (сухое коровье молоко, сгущенное молоко с сахаром, сливочное масло, маргарин, яичный порошок и т.д.), а также готовые хлебобулочные изделия.
Наиболее разнообразное применение из гравиметрических методов получили методы осаждения. Напомним (см. раздел 2.1), что они основаны на том, что определяемый компонент количественно осаждают химическими способами (т.е. при взаимодействии с подходящим реактивом-осадителем) в виде малорастворимого соединения, ПР которого не превышает  . Выделившийся осадок отделяют, промывают, высушивают, прокаливают (если нужно) и взвешивают.
. Выделившийся осадок отделяют, промывают, высушивают, прокаливают (если нужно) и взвешивают.
В настоящем пособии подробно рассмотрены теоретические основы и особенности только методов осаждения.
3.3. Основные операции гравиметрического анализа.
Осаждаемая и гравиметрическая формы осадка
Г.а. методом осаждения проводят в следующей последовательности:
1) отбирают среднюю пробу анализируемого образца;
2) взвешивают навеску для анализа;
4) осаждают определяемый компонент;
5) отделяют осадок фильтрованием;
10) вычисляют результат анализа.
Заметим, что прокаливание осадков в некоторых случаях невозможно (см. раздел 3.5.9), и тогда эта стадия исключается.
Поскольку многие осадки при прокаливании изменяют свой состав, то различают осаждаемую и гравиметрическую (весовую) формы осадка.
Осаждаемая форма осадка (форма осаждения) — соединение, которое осаждается из раствора при взаимодействии определяемого компонента с соответствующим реагентом (или соединение, в виде которого осаждают анализируемое вещество).
Гравиметрическая (весовая) форма — соединение, которое взвешивают для получения окончательного результата анализа (или соединение, в виде которого взвешивают определяемый компонент).
Наиболее часто составы осаждаемой и гравиметрической форм не совпадают. Реже их состав одинаков. Например, хлорид- и Ag + -ионы определяют г.а., осаждая их в виде AgCl.
Осаждаемая и гравиметрическая формы в этом случае имеют одинаковый состав AgCl.
3.4. Требования к осаждаемой и гравиметрической формам осадка
Для получения точных результатов в гравиметрических методах осаждаемая и гравиметрическая формы осадка должны соответствовать определенным требованиям.
Требования к осаждаемой форме:
1. Осадок должен быть практически нерастворим.
Это означает, что в растворе после осаждения и промывания осадка, определяемого компонента должно оставаться меньше, чем можно взвесить на аналитических весах. Опыт показывает, что для бинарных электролитов (т.е. соединений, каждая молекула которых образует при диссоциации два иона, например, AgI, PbSO4 и т.д.) практически полное осаждение может быть достигнуто лишь тогда, когда ПР осадка имеет значение, не превышающее . (Если же соединение тринарное, тетрарное и более сложного состава, то ПР будет другим, но всегда растворимость осадка не должна превышать 10 -6 г/л).
2. Желательно, чтобы структура осадка давала возможность с достаточной скоростью выполнить операции фильтрования и промывания осадка.
В этом отношении очень удобны крупнокристаллические осадки, т.к.они почти не забивают поры фильтра, мало загрязняются посторонними примесями из анализируемого раствора и легко отмываются от них.
Аморфные осадки типа Al(OH)3 и Fe(OH)3, имея сильно развитую поверхность, являясь рыхлыми и объемными, значительно адсорбируют примеси из анализируемого раствора, трудно отмываются от них, а также медленно фильтруются. Однако, если соединений, обладающих более подходящими для анализа свойствами, не известно, то работают и с такими осадками. Стремятся только обеспечить все условия, способствующие наибольшему устранению или уменьшению недостатков аморфных осадков (эти условия представлены в разделе 3.5.5).
3. Необходимо, чтобы осаждаемая форма достаточно легко и полностью превращалась в гравиметрическую форму.
Требования к гравиметрической форме:
1. Состав гравиметрической формы должен соответствовать определенной химической формуле.
На практике многие осадки не удовлетворяют этому требованию. Например, осадок гидроксида железа не соответствует формуле Fe(OH)3, более правильно принято писать 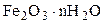 . Количество воды в этом соединении непостоянно и зависит от условий осаждения. Но после прокаливания гидроксид железа переходит в стабильную гравиметрическую форму, строго соответствующую формуле Fe2O3.
. Количество воды в этом соединении непостоянно и зависит от условий осаждения. Но после прокаливания гидроксид железа переходит в стабильную гравиметрическую форму, строго соответствующую формуле Fe2O3.
2. Гравиметрическая форма должна обладать достаточной химической устойчивостью.
Иначе нарушится соответствие ее состава определенной химической формуле. Если установлено, что гравиметрическая форма может легко изменять свой состав вследствие, например, поглощения водяных паров или СО2 из воздуха, окисления (восстановления), разложения или других подобных реакций, то ее превращают в более удобную форму, обрабатывая соответствующими ре-агентами. Например, осадок СаО, легко поглощающий Н2О и СО2 из воздуха (что затрудняет его точное взвешивание), иногда превращают в CaSO4, обработав серной кислотой.
3. Гравиметрическая форма, получаемая путем прокаливания, должна обладать устойчивостью при высоких температурах (термоустойчивостью).
Некоторые гравиметрические формы могут разлагаться при высоких (порядка 1200-1300 о С и выше) температурах.
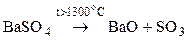 ;
;
 ,
,
поэтому осадки прокаливают при температуре не выше 1100 о С.
4. Молекулярная масса гравиметрической формы должна быть по возможности большей.
Иными словами, содержание определяемого элемента в гравиметрической форме должно быть как можно меньшим. Благодаря этому, относительная погрешность определения в меньшей мере влияет на результат анализа.
3.5. Техника гравиметрического анализа. Выбор оптимальных условий выполнения гравиметрического определения
3.5.1. Отбор средней пробы анализируемого образца
Понятие «средняя проба» и правила ее отбора для анализа представлены в разделе 1.7.5. Кроме средней, известно еще несколько видов проб. Описание основных из них приведено в «Аналитической химии» [16].
Обычно, отобрав представительную первичную пробу анализируемого продукта, ее измельчают (в случае твердых и сыпучих материалов), перемешивают и сокращают до размеров лабораторной (паспортной) пробы. Из нее отбирают аналитические пробы для проведения отдельных анализов.
3.5.2. Взятие навески для анализа
Навеску анализируемого образца рассчитывают с учетом массы гравиметрической формы. Опытным путем установлено, что оптимальная масса гравиметрической формы для кристаллических осадков составляет около 0,5 г, для аморфных — 0,1 г.
Расчет исходной навески для анализа ведут, исходя из уравнений реакций, протекающих при гравиметрическом определении. Причем, если анализируемое вещество содержит значительное количество примесей, то навеска должна соответствовать только содержанию определяемого компонента в исследуемом образце.
Например, если анализируют 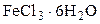 на содержание Fe 3+ -ионов, то навеску (mи.с.) вычисляют, исходя из следующей схемы.
на содержание Fe 3+ -ионов, то навеску (mи.с.) вычисляют, исходя из следующей схемы.
В общем виде расчет mи.с. осуществляют по формулам:
а) при осаждении кристаллических осадков:
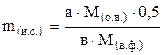 ; (40)
; (40)
б) при осаждении аморфных осадков:
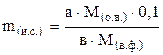 , (41)
, (41)
где а — стехиометрический коэффициент в уравнении реакции перед определяемым веществом;
в — стехиометрический коэффициент в уравнении реакции перед соединением весовой формы.
Рассчитанную навеску сначала взвешивают ориентировочно на технических весах, а затем — на аналитических весах.
Для взятия навесок твердых веществ обычно применяют часовые стекла, бюксы (допускается калька), а для жидких веществ — капельницы, колбы емкостью 1-2 см 3 и т.д. Многолетней практикой созданы наиболее рациональные способы перенесения навески анализируемого вещества в стакан. Они подробно рассмотрены, например, в «Основах аналитической химии» [8].
В качестве растворителей в гравиметрии применяют:
— растворы кислот или их смеси, например, «царскую водку» (смесь 1-го объема концентрированной HNO3 с тремя объемами концентрированной HCl);
Выбор подходящего из перечисленных растворителя делают до гравиметрического определения путем качественных проб.
а) характер реакции, происходящей при действии реактива-растворителя;
б) растворимость образующихся в результате соединений;
в) дальнейший ход анализа исследуемого образца.
Например, для растворения CaCO3 удобнее использовать HCl, а не H2SO4, т. к. CaSO4 малорастворим в воде. При анализе природных сернистых руд целесообразно применять окислители, переводящие серу в  -анион и т.д.
-анион и т.д.
Чаще всего в гравиметрии в качестве растворителей применяют дистиллированную воду, холодную или нагретую до определенной температуры.
Растворение навески ведут в фарфоровых стаканах или колбах, реже — в фарфоровых чашках. Растворение в кислотах (разбавленных или концентрированных) выполняют в вытяжном шкафу.
Если анализируемый образец имеет очень сложный состав (т.е. состоит из большого числа компонентов), то подобрать подходящий растворитель трудно. В таких случаях для разложения вещества и перевода определяемой составной части в раствор прибегают к сплавлению (или «спеканию») вещества с теми или иными «плавнями» (например, K2S2O7, KHSO4 и др.). Подробнее об этом представлено в работе В.Н. Алексеева [1].
3.5.4. Осаждение. Выбор и количество осадителя
Эта стадия является одной из наиболее важных в г.а. От качества, формы, структуры и степени чистоты получаемых осадков в значительной степени зависят точность и надежность результатов анализа. В свою очередь, структура и свойства образующихся осадков зависят от множества факторов: их растворимости, концентрации осаждаемого вещества, осадителя, скорости осаждения, рН-среды и т.д. Важное значение имеет также правильный выбор осадителя-ре-агента. Его делают, исходя из требований к осаждаемой и гравиметрической формам осадка (см. раздел 3.4) с учетом следующих соображений:
1. Осадитель должен быть таким, чтобы ПР образующегося осадка не превышало и при этом было как можно меньшим.
Например, ионы Ва 2+ образуют осадки с (NH4)2C2O4 ( 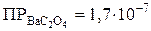 ); (NH4)2CO3 (
); (NH4)2CO3 ( 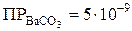 ); K2CrO4 (
); K2CrO4 ( 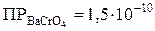 ); и с H2SO4 (
); и с H2SO4 ( 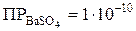 ).
).
Очевидно, что при гравиметрическом определении ионов Ва 2+ в качестве осадителя наиболее целесообразно взять H2SO4 или K2CrO4.
В гравиметрии в качестве осадителей часто применяют реактивы, позволяющие получать гидроксиды, карбонаты, сульфаты, сульфиды, оксалаты, фосфаты металлов.
2. Желательно, чтобы осадитель был летучим веществом.
Опыт показывает, что при выделении осадка из раствора он всегда увлекает растворенные посторонние вещества или ионы, в том числе и ионы осадителя. От этих примесей осадок приходится отмывать, и это не всегда удается сделать полностью. Но, благодаря летучести, оставшаяся при промывании часть осадителя легко удалится при прокаливании и не станет причиной ошибки анализа, поэтому часто для осаждения применяют соединения аммония: гидроксид, карбонат, оксалат и т.д.
3. Осадитель должен быть специфическим, т.е. осаждать только определяемый ион.
Это важно, т.к. при выполнении большинства анализов определяемый ион приходится осаждать в присутствии ряда других ионов.
Заметим, что специфический осадитель удается найти не всегда, поэтому проводят маскировку мешающих определению ионов, т.е. связывают их в достаточно прочные комплексы, не осаждаемые выбранным реактивом-осади-телем. Известны и другие способы удаления мешающих ионов из раствора [1].
Количество осадителя. Для уменьшения потерь от растворимости осадка в гравиметрии к анализируемому раствору добавляют избыток осадителя. Обычно осадителя берут в 1,5 раза больше, чем рассчитано по уравнению реакции осаждения. Иногда, если это необходимо, прибавляют значительно боль-шее (в 2-3 раза) количество осадителя. Однако слишком большой избыток осадителя вреден, т.к. он вызывает не понижение, а наоборот — повышение растворимости осадка. Причиной этого может быть образование растворимых комплексных соединений или кислых солей, амфотерность осаждаемого соединения (гидроксида), «солевой эффект» (вследствие возрастания ионной силы раствора) и т.д.
Необходимый объем осадителя рассчитывают по формуле:
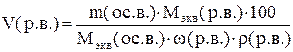 , (42)
, (42)
где ос.в. — осаждаемое вещество;
Этот расчет ориентировочный, поэтому полученное значение объема округляют до одной-двух значащих цифр. Рассчитанный объем раствора осадителя отмеривают с точностью до 0,1-0,2 см 3 .
3.5.5. Осаждение. Оптимальные условия осаждения кристаллических и аморфных осадков
В зависимости от индивидуальных свойств анализируемых веществ и условий осаждения процесс образования осадка может идти двумя путями, которые приводят к образованию либо кристаллического, либо аморфного осадка. В разделе 3.4 было отмечено, что в гравиметрии отдают предпочтение крупнокристаллическим осадкам. Прежде, чем указать условия, при которых они получаются, необходимо хотя бы в общих чертах рассмотреть, как протекает процесс образования осадка (твердой фазы) в растворе.
Механизм образования кристаллических и аморфных осадков. Процесс образования осадка (твердой фазы) в растворе очень сложен. Сначала почти всегда наблюдается так называемый индукционный период, который длится от момента смешения растворов анализируемого вещества и осадителя до появления видимого осадка.
Индукционный период может быть очень большим (например, при осаждении BaSO4) или совсем непродолжительным (при образовании AgCl). Наличие этого периода объясняется тем, что образование осадка проходит ряд стадий. По мере прибавления в анализируемый раствор реактива-осадителя наступает момент, когда произведение концентраций ионов осаждаемого соединения превысит ПР осадка и образуется осадок малорастворимого соединения сначала в виде зародышевых, или первичных кристаллов. Для образования таких кристаллов:
а) в растворе должно столкнуться в определенном соотношении и при определенном расположении довольно большое число реагирующих ионов;
б) гидратная (сольватная) оболочка ионов должна быть разрушена.
На стадии образования первичных кристаллов не наблюдается еще видимого выделения вещества в осадок, т. к. сами эти кристаллы, а также их агрегаты (агрегация — процесс соединения зародышевых кристаллов в более крупные, состоящие из десятков, сотен молекул) имеют очень малый размер, поэтому эта стадия формирования осадка соответствует существованию коллоидных систем. Затем первичные кристаллы или их агрегаты образуют более крупные частицы и выпадают в осадок. Этот процесс в зависимости от индивидуальных свойств анализируемых веществ может идти двумя путями, определяющими форму осадка, т.е. его образование в виде кристаллов или в аморфном состоянии.
1. Если растворимость осадка не слишком мала, то образуется кристаллический осадок. При медленном прибавлении в раствор новых порций осадителя не появляются новые центры кристаллизации (первичные кристаллы) или новые агрегаты. Раствор некоторое время остается в пересыщенном состоянии. Выделение вещества из этого пересыщенного раствора происходит преимущественно на поверхности ранее образовавшихся зародышевых кристаллов, которые постепенно растут. Кроме того, первичный осадок непрерывно растворяется и по достижении границы пересыщения вновь выпадает. Это ведет к растворению мелких и росту крупных кристаллов. В конечном итоге получается кристаллический осадок, состоящий из сравнительно небольшого числа относительно крупных кристаллов.
2. Если растворимость осадка мала, то образуется аморфный осадок. Прибавление каждой новой порции осадителя вызывает быстрое возникновение в растворе огромного количества мельчайших зародышевых кристаллов, которые растут уже не вследствие отложения на их поверхности соответствующего вещества, а в результате их соединения в более крупные агрегаты, оседающие под влиянием силы тяжести на дно сосуда. Происходит коагуляция первоначально образующегося коллоидного раствора, образуется аморфный осадок.
Относительное пересыщение. Выпадению осадка предшествует пересыщение раствора. В пересыщенном растворе образуются зародышевые кристаллы осаждаемого вещества. Скорость образования этих центров кристаллизации и их число зависят от степени пересыщения раствора.
Для характеристики пересыщения часто применяют формулу:
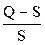 , (43)
, (43)
где Q = С/2, С — молярная концентрация каждого из реагентов;
S — растворимость малорастворимого вещества.
Формула является эмпирической и справедливой только при одинаковой концентрации реагирующих веществ.
Отношение называют относительным пересыщением. Им пользуются для определения оптимальных условий осаждения кристаллических и аморфных осадков.
Оптимальные условия получения кристаллических осадков. Из формулы (43) следует, что, если относительное пересыщение мало (значение Q мало, а S велико), то меньшее число первичных кристаллов будет возникать, и крупнее они будут. Следовательно, для получения крупнокристаллических осадков необходимо в процессе осаждения повышать растворимость осадка и понижать концентрации осаждаемого и осаждающих ионов.
Для повышения растворимости (S) осадков в процессе осаждения:
а) обычно повышают температуру: ведут осаждение из горячего анализируемого раствора, иногда нагревают и раствор осадителя; но осадок отфильтровывают только после охлаждения раствора;
б) добавляют в раствор вещества, повышающие растворимость осадков.
Заметим, что после того, как осадок сформируется, растворимость понижают, иначе осаждение будет неполным. Например, при осаждении сульфата бария прибавляют HCl, которая повышает его растворимость вследствие образования 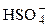 -ионов. К концу осаждения эту повышенную растворимость BaSO4 снова понижают, прибавляя небольшой избыток осадителя.
-ионов. К концу осаждения эту повышенную растворимость BaSO4 снова понижают, прибавляя небольшой избыток осадителя.
Для понижения концентрации реагирующих ионов при формировании осадка: а) ведут осаждение из разбавленного анализируемого раствора разбавленным раствором осадителя; б) раствор с осадком непрерывно перемешивают;
Перемешивание способствует снижению высоких местных (локальных) концентраций осадителя и увеличению скорости растворения первичного осадка. Число зародышевых кристаллов при этом уменьшается, и создаются условия для роста крупных кристаллов.
в) осадитель добавляют очень медленно, по каплям (особенно в начале осаждения);
При медленном осаждении (наряду с одновременным тщательным перемешиванием анализируемого раствора) обеспечивается равномерное распределение и малая концентрация осадителя во всем объеме раствора. Вследствие этого степень пересыщения невелика и первичных центров кристаллизации появляется сравнительно немного. Образующиеся первичные кристаллы успевают правильно ориентироваться по отношению друг к другу (ориентация — расположение частиц осадка в процессе агрегации в строго определенном порядке). Дальнейшее прибавление новых порций осадителя приводит не столько к возникновению новых центров кристаллизации, сколько к увеличению размеров уже образовавшихся центров и к получению крупнокристаллических осадков. Чем больше скорость образования осадка, тем быстрее нарушается правильная ориентация при кристаллизации. При быстром осаждении сразу появляется много зародышевых, а затем мелких кристаллов.
г) иногда связывают осаждаемый ион в комплексное соединение средней прочности;
Достаточно низкая концентрация осаждаемого иона в растворе создается за счет частичной диссоциации комплексного соединения. При добавлении оса-дителя концентрация осаждаемого иона понижается из-за образования осадка, а, следовательно, будет смещаться равновесие ионизации комплекса. В раствор переходят новые ионы о.в., но их концентрация все время будет оставаться низкой.
д) применяют возникающие реагенты.
В раствор добавляют не осадитель, а вещество, которое, вступая в какую-либо реакцию, образует этот осадитель. Необходимым условием является медленное протекание такой реакции, в результате чего ион осадителя постепенно, очень маленькими порциями «возникает» (образуется) в исследуемом растворе. Его концентрация всегда остается низкой, что способствует понижению относительного пересыщения раствора. Например, существуют реагенты, которые претерпевают в растворе медленный гидролиз с выделением собственно осадителя. К таким реагентам относятся некоторые сложные эфиры: триэтилфосфат, диметилфосфат, метилоксалат, этилоксалат и т.д. Один из них, диметилсульфат, применяют для гравиметрического определения ионов Ва 2+ . Ионы Ва 2+ осаждаются H2SO4, образующейся в результате медленного гидролиза диметилсульфата.
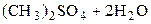
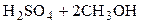
Такой метод называется методом возникающих реагентов. Процесс осаждения с помощью возникающих реагентов называют также процессом осаждения из гомогенного раствора.
Оптимальные условия получения аморфных осадков. Из формулы (43) видно, что если вещество малорастворимо (величина S мала) и выделяется из сравнительно концентрированных растворов (Q велико), то относительное пересыщение велико. Это будет способствовать быстрому образованию в раст-воре огромного количества зародышевых кристаллов, их быстрой агрегации и выделению аморфного осадка, поэтому первым условием при получении аморфных осадков является следующее: их необходимо осаждать из концентрированных растворов концентрированным раствором осадителя. Таким способом, они получаются гораздо более плотными, с меньшей поверхностью, быстрее оседают и легче отмываются от примесей.
Другие условия получения аморфных осадков обусловлены особенностями их образования. Ранее уже было отмечено, что в результате соединения первичных кристаллов в более крупные агрегаты происходит коагуляция коллоидного раствора, выпадает аморфный осадок. Очевидно, что при получении такого осадка наиболее важно создать условия, способствующие быстрому про-теканию именно этого процесса. Они определяются необходимостью устранить влияние двух основных факторов, препятствующих коагуляции коллоидных частиц и укрупнению частиц коллоидных осадков.
Первым фактором, мешающим сцеплению коллоидных частиц друг с другом, является наличие у них одноименных электрических зарядов, между которыми действуют силы электростатического отталкивания (эти заряды возникают в результате адсорбции коллоидными частицами ионов из раствора) (см. раздел 3.5.7). Для того, чтобы устранить его негативное влияние, осаждение аморфных осадков ведут в присутствии подходящего электролита-коагу-лянта. Заряд иона этого электролита всегда противоположен знаку заряда частиц, поэтому ионы электролита-коагулянта, адсорбируясь на поверхности коллоидных частиц, нейтрализуют их заряд и дают им возможность соединяться в агрегаты. В качестве электролитов-коагулянтов в гравиметрии обычно применяют различные соли аммония или кислоты.
Другим фактором, препятствующим сцеплению коллоидных частиц, является адсорбция ими молекул растворителя — сольватация коллоидных частиц (если растворителем является вода, то говорят о гидратации коллоидных частиц). Сольватные оболочки мешают коллоидным частицам достаточно близко подойти друг к другу и объединиться в более крупные агрегаты.
Разрушение сольватных оболочек достигается двумя путями:
а) прибавлением раствора электролита-коагулянта высокой концентрации;
б) повышением температуры раствора, из которого ведут осаждение.
В первом случае ионы электролита-коагулянта «отнимают» молекулы растворителя у коллоидных частиц (при этом сами сольватируются) и одновременно нейтрализуют их заряд. Во втором случае нагревание раствора не только способствует разрушению сольватных оболочек коллоидных частиц, но одновременно уменьшает адсорбцию загрязняющих осадок ионов (см. раздел 3.5.7), поэтому аморфные осадки получают из горячего раствора.
Обобщая изложенное об условиях получения аморфных осадков, отметим, что выбор оптимальных из них определяется тем, что необходимо:
а) сделать быстрым и полным процесс коагуляции первоначально образующегося коллоидного раствора;
б) предупредить явление адсорбции осадков загрязняющими ионами из раствора.
Оптимальные условия осаждения аморфных осадков суммированы в табл. 3 (см. стр. 53).
Старение (созревание) осадков. Под старением (созреванием) осадка понимают все необратимые структурные изменения, происходящие в осадке при его выдерживании в течение различного времени под маточным раствором. Маточный раствор — раствор над осадком после завершения стадии осаждения.
Для кристаллических осадков старение — очень важная стадия, длящаяся несколько часов (обычно до следующего дня). Когда осадок находится под маточным раствором, происходит ряд процессов, приводящих к укрупнению и совершенствованию кристаллов, получению их в чистой, практически свободной от примесей форме. Причиной укрупнения кристаллов является более высокая растворимость очень мелких кристаллов вещества по сравнению с растворимостью более крупных кристаллов. Подобное увеличение растворимости с умень-шением размеров кристаллов объясняется поверхностным натяжением, стремящимся возможно сильнее уменьшить поверхность соприкосновения раствора с осадком. В результате мелкие кристаллы растворяются, затем растворенное вещество отлагается на поверхности крупных кристаллов, и последние посте-пенно растут.
Этот процесс ускоряется: а) при повышении температуры раствора; б) пе-ремешивании раствора.
При старении кристаллического осадка происходит также совершенствование формы кристаллов. Причиной этого является то, что при выдерживании осадка под маточным раствором между ними устанавливается динамическое равновесие. Одни ионы переходят в раствор с поверхности осадка, другие — осаждаются на поверхности кристаллов осадка. Ионы кристаллической решетки, находящиеся на «несовершенных» местах поверхности осадка, переходят в раствор и затем осаждаются в узлах более «совершенного» кристалла. Происходит перекристаллизация осадка. Совершенствование формы кристаллов способствует повышению чистоты осадка.
Аморфные осадки не выдерживают под маточным раствором (не оставляют для созревания). Оставлять аморфные осадки в соприкосновении с раствором вредно, т.к. они:
а) даже при длительном выдерживании сохраняют студенистый вид и очень большую поверхность, и поэтому сильно загрязняются ионами, адсорбированными из раствора;
б) нередко при старении переходят в другие, менее растворимые модификации.
Аморфные осадки после осаждения сразу же! подвергают дальнейшим операциям: переносят на фильтр и промывают.
Техника осаждения. Обычно осаждение ведут в том же сосуде (химическом стакане), в котором проводилось растворение пробы. Осаждение кристаллических и аморфных осадков проводят с учетом условий, перечисленных в табл. 3. Осадки получают при нагревании растворов одного или обоих реагирующих веществ. При этом не следует нагревать до кипения, так как может произойти потеря вещества вследствие разбрызгивания. Осадитель обычно добавляют из бюретки или пипетки, а также из химического стакана с помощью стеклянной палочки. (Заметим, что на одном конце стеклянной палочки должно быть узкое резиновое кольцо, плотно прилегающее к ней. Во время осаждения палочку опускают в стакан именно этим концом.)
При добавлении осадителя всегда стремятся к тому, чтобы его раствор стекал по внутренней стенке стакана или по стеклянной палочке, а не падал каплями в середину стакана, что может привести к разбрызгиванию. Для осаждения кристаллических осадков пользуются разбавленными растворами осадителя, поэтому отмеренный объем (или отвешенное количество) осадителя разбавляют водой примерно до 50 см 3 . Для осаждения аморфных осадков пользуются концентрированными растворами осадителя, поэтому отмеренный объем осадителя разбавляют водой только до 5 см 3 . Во время осаждения раствор перемешивают стеклянной палочкой, следя за тем, чтобы палочка касалась дна и стенок стакана, но не царапала стекло. Каждый раз, когда палочку вынимают из стакана, ее следует промывать дистиллированной водой над стаканом.
После добавления рассчитанного количества осадителя всегда проверяют полноту осаждения. Для этого дают осадку собраться на дне стакана, и, когда жидкость над стаканом посветлеет, добавляют несколько (обычно 2-3) капель раствора осадителя. Отсутствие помутнения в месте падения капель указывает на полноту осаждения.
Осажденный кристаллический осадок оставляют на некоторое время (1-6 часов, иногда до следующего дня) для созревания.
Когда заканчивается осаждение аморфного осадка, в стакан прибавляют 100-150 см 3 горячей дистиллированной воды и быстро фильтруют, а затем промывают.
Оптимальные условия осаждения
| Кристаллических осадков | Аморфных осадков |
| Осаждение проводят: | |
| 1) из достаточно разбавленных анализируемых растворов разбавленным раствором осадителя; | 1) из концентрированных анализируемых растворов концентрированным раствором осадителя; |
| 2) из горячего раствора (иногда нагревают и раствор осадителя). | |
| Добавляют осадитель: | |
| 3) очень медленно, по каплям (особенно в начале осаждения); | 3) быстро! |
| 4) непрерывно перемешивая раствор стеклянной палочкой во избежание сильных местных пересыщений при добавлении осадителя; | |
| 5) в присутствии электролита, | |
| повышающего растворимость осадка; | коагулянта. |
| После окончания осаждения: | |
| 6) осадок оставляют для созревания (старение осадка); | 6) сразу же по окончании осаждения к раствору с осадком приливают большой объем ( 100-150 см 3 ) горячей дистиллированной воды и смесь перемешивают; 7) осадок не оставляют для созревания, а немедленно переносят на фильтр и промывают. |
После переведения определяемого компонента в осадок, последний отделяют от маточного раствора фильтрованием. Перед фильтрованием раствор с осадком охлаждают для уменьшения растворимости осадка.
а) через стеклянные фильтры (фильтры Шотта, стеклянные фильтрующие тигли); они представляют собой небольшие стеклянные сосуды с впаянной внутри пористой стеклянной пластинкой; применяются для отфильтровывания крупнокристаллических осадков;
б) тигли Гуча (в них фильтром является слой волокнистого асбеста, помещаемый на сетчатое дно тигля);
в) фарфоровые тигли с пористым дном (в отличие от стеклянных тиглей они выдерживают нагревание до очень высокой температуры);
г) бумажные беззольные фильтры (эти фильтры применяют при фильтровании мелкокристаллических и аморфных осадков).
Бумажные фильтры являются беззольными, т.к. они сгорают почти полностью. Масса остающейся при этом золы составляет 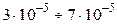 г (т.е. за пределом точности взвешивания на аналитических весах). Если же масса золы превышает 0,0002 г, то ее значение вычитают из массы осадка.
г (т.е. за пределом точности взвешивания на аналитических весах). Если же масса золы превышает 0,0002 г, то ее значение вычитают из массы осадка.
Беззольные фильтры различаются по диаметру (6, 7, 9 и 11 см) и по плотности (пористости) фильтровальной бумаги. Различие по плотности определяется по цвету бумажной ленты, которой оклеивают упаковку готовых фильтров. Приняты следующие условные обозначения:
— красная (или черная) лента — быстрофильтрующие фильтры (диаметр пор
— белая лента — бумага средней проницаемости (диаметр пор
 м);
м);
— синяя лента — «баритовые», плотные фильтры (диаметр пор
 м);
м);
— желтая лента — обезжиренные фильтры.
Аморфные осадки фильтруют через фильтры малой плотности (красная или черная лента), кристаллические — средней и большой плотности (белая или синяя лента).
При выполнении процесса фильтрования учитывают следующие правила:
1. Выбирая размер фильтра, руководствуются не объемом фильтруемой жидкости, а количеством осадка. Осадок не должен занимать больше 1/3-1/2 фильт-тра, иначе его будет невозможно хорошо промыть. Не следует брать и фильт-ры слишком больших размеров.
2. Размер воронки подбирают так, чтобы края фильтра были на 5-15 мм ниже края воронки.
3. До фильтрования фильтр смачивают дистиллированной водой и осторожно! прижимают (прилаживают, подгоняют) к воронке так, чтобы между ними не было пузырьков воздуха.
4. Воронку с фильтром помещают в кольцо штатива, подставив под нее стакан для слива так, чтобы скошенный конец трубки воронки касался стенки стакана (рис.4). Этим предотвращают разбрызгивание жидкости при фильтровании.
5. До начала фильтрования носик стакана с осадком полезно слегка потереть пальцем с наружной стороны. Благодаря этому простому приему, он не будет смачиваться водой, и капли фильтруемой жидкости не будут стекать по внешней стенке стакана.
6. Фильтрование проводят следующим способом.
Стеклянную палочку, которой перемешивали раствор в процессе осаждения, вынимают из стакана и держат левой рукой в вертикальном положении над воронкой. Нижний конец палочки (с резиновым наконечником) должен подходить к фильтру, не касаясь бумаги, близко от той части фильтра, где он сложен втрое. Затем правой рукой берут стакан с осадком, прикладывают носик стакана к палочке и осторожно сливают жидкость на фильтр (рис. 4). По мере наполнения фильтра палочку поднимают так, чтобы она не касалась жидкости. Фильтр нельзя наполнять жидкостью до краев, только на 2/3 (т.е. уровень жидкости должен быть не меньше, чем на 5 мм ниже края фильтра). Налив жидкость на фильтр, медленно приводят стакан в вертикальное положение, ведя его носик по стеклянной палочке кверху (этим предотвращается стекание последней капли жидкости по внешней стенке стакана). При фильтровании стеклянная палочка должна находиться либо над фильтром, либо в стакане. Класть палочку на стол нельзя!, т.к. при этом будут потеряны оставшиеся на ней частицы осадка.
7. Фильтрование продолжают до тех пор, пока еще можно сливать жидкость с осадка, затем, убедившись в прозрачности фильтрата, осадок промывают.
Промывание осадков необходимо для удаления адсорбированных на поверхности осадка примесей, а также маточного раствора, пропитывающего осадок. Кроме того, при промывании удаляются такие соли, которые не улетучиваются при прокаливании.
Адсорбция — одна из причин загрязнения осадков. Адсорбция — это поглощение примесей (ионов или молекул) из раствора поверхностью частиц осадка.
1. Бюкс 2. Тигли:
Дата добавления: 2016-11-23 ; просмотров: 9061 | Нарушение авторских прав
источник