Свинец ядовит и обладает кумулятивными свойствами (способностью накапливаться в организме). Вследствие этого наличие свинца во всех видах консервов не допускается.
Основные источники попадания свинца в консервы – полуда, содержание свинца в которой ограничивается до 0,04 %, и припой. Наличие в консервируемых продуктах веществ, способных растворять металлы, может привести при длительном хранении консервов к переходу свинца в состав содержимого банки. Содержание свинца в продукте определяют в случае длительного хранения и наличия на внутренней стороне банки наплывов припоя.
Метод основан на получении раствора хлорида свинца после озоления навески продукта, осаждении из раствора сульфидов металлов и определении свинца в насыщенном растворе ацетата натрия в присутствии бихромата калия.
Порядок выполнения анализа: 15 г измельченного продукта помещают в фарфоровую чашку диаметром около 7 см, высушивают на песочной бане или в сушильном шкафу, а затем осторожно обугливают и озоляют на слабом огне или муфельной печи при слабо-красном накаливании стенок муфеля. К золе добавляют 5 мл разбавленной соляной кислоты (соотношение1:1), 1 каплю пероксида водорода и выпаривают на водяной бане досуха. К сухому остатку добавляют 2 мл 10 %-ной соляной кислоты и 3 мл воды, после чего содержимое чашки фильтруют через предварительно смоченный водой фильтр в коническую колбу вместимостью 100 мл. Чашку и фильтр промывают 15 мл дистиллированной воды, собирая промывные воды в ту же колбу. Полученный раствор нагревают до 40-50 ˚С, пропуская через него в течение 40 — 60 минут сероводород через узко оттянутую трубку, доходящую до дна колбы. При этом в осадок выпадают сульфиды свинца, олова, меди. Выпавший осадок сульфидов и серы отделяют центрифугированием в пробирке вместимостью 10 мл. Жидкость сливают, а осадок сульфидов металлов промывают 1 – 2 раза 1%-ным раствором соляной кислоты, насыщенным сероводородом. К промытому осадку сульфидов тотчас же добавляют 5 капель 10 %-ного раствора гидроксида натрия (во избежание окисления сульфида свинца в сульфат, растворимый в щелочах), нагревают на кипящей водяной бане, вводят 10 мл воды и центрифугируют. При большом осадке обработку гидроксидом натрия производят два раза.
К осадку сульфидов свинца и меди добавляют 5 – 10 капель смеси крепкой серной и азотной кислоты, взятых в равных количествах, осторожно нагревают на небольшом пламени горелки до полного удаления паров азотной кислоты и появления белых густых паров триоксида серы. После охлаждения в пробирку добавляют 0,5 –1,5 мл дистиллированной воды и такое же количество этанола. Если после прибавления воды и спирта раствор остается прозрачным, то соли свинца считают необнаруженными. При появлении в растворе мути или выпадения белого осадка сульфат свинца отделяют разбавленным этанолом (соотношение 1:1). К оставшемуся в центрифужной пробирке осадку сульфата свинца добавляют 1 мл насыщенного раствора ацетата натрия, предварительно слабо подкисленного уксусной кислотой, и нагревают на кипящей водяной бане 5 – 10 минут. Затем приливают 1 мл дистиллированной воды, после чего содержимое пробирки фильтруют через маленький фильтр, смоченный дистиллированной водой. Фильтрат собирают в мерный цилиндр вместимостью 10 мл. Пробирку и фильтр промывают несколько раз, небольшими порциями дистиллированной воды, собирая промывные воды в тот же цилиндр. Объем раствора доводят водой до метки и перемешивают. 5 мл раствора из цилиндра переносят в центрифужную пробирку, добавляют 3 капли 5%-ного раствора бихромата калия и перемешивают. Если раствор остается прозрачным в течение 10 мин, считают, что свинец не обнаружен. При наличии свинца в растворе появляется желтая муть (PbCrO4). В этом случае проводят количественное определение свинца.
Для количественного определения свинца определенный объем раствора (0,5 – 2 мл) из цилиндра переносят в плоскодонную пробирку с делениями на 10 мл. В три другие такие же пробирки вносят стандартный раствор с содержанием свинца 0,01; 0,015 и 0,02 мг. В пробирки со стандартным раствором добавляют такое количество насыщенного раствора ацетата натрия, слабо подкисленного уксусной кислотой, чтобы его содержание в испытуемом и стандартном растворах было одинаковым (если для количественного определения свинца берут 1 мл испытуемого раствора, то в пробирки со стандартным раствором свинца добавляют 0,1 мл ацетата натрия). Далее во все четыре пробирки добавляют дистиллированную воду до 10 мл, перемешивают и приливают по 3 капли 5%-ного раствора бихромата калия. Содержимое пробирки хорошо перемешивают и через 10 – 15 минут мутность испытуемого раствора сравнивают с мутностью стандартных растворов.
Содержание свинца определяют по формуле 6.
где х – содержание свинца в 1 кг продукта, мг;
а – количество свинца в пробирке стандартным раствором, мг;
V – объем раствора, взятый для сравнения со стандартным раствором, мл; 15 – навеска продукта, г.
Приготовление стандартного раствора нитрата свинца. 160 мг нитрата свинца растворяют в небольшом количестве дистиллированной воды в мерной колбе вместимостью 100 мл, добавляют 1 каплю концентрированной азотной кислоты, перемешивают и доводят объем дистиллированной водой до метки; 1 мл такого раствора содержит 1 мг свинца, 2 мл раствора переносят в мерную колбу вместимостью 100 мл, доводят объем дистиллированной водой до метки. Последний раствор является стандартным. В 1 мл его содержится 0,02 мг свинца.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: Студент — человек, постоянно откладывающий неизбежность. 10008 —  | 7152 —
| 7152 —  или читать все.
или читать все.
источник
Настоящий документ устанавливает фотометрическую методику количественного химического анализа проб природных и очищенных сточных вод для определения в них ионов свинца при массовой концентрации от 0,002 до 0,03 мг/дм 3 .
Если массовая концентрация ионов свинца в анализируемой пробе превышает верхнюю границу диапазона, то допускается разбавление пробы таким образом, чтобы концентрация ионов свинца соответствовала регламентированному диапазону.
Мешающие влияния, обусловленные присутствием в пробе органических веществ, ионов меди, цинка, кадмия, никеля, серебра, ртути, висмута устраняются специальной подготовкой пробы к анализу (п. 9).
Фотометрический метод определения массовой концентрации ионов свинца основан на взаимодействии свинца с дифенилтиокарбазоном (дитизоном) в четыреххлористом углероде с образованием комплексного соединения, окрашенного в красный цвет. Оптическую плотность раствора комплексного соединения измеряют при l = 520 нм.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведённых в таблице 1.
Значения показателей точности, повторяемости и воспроизводимости методики
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ± d , %
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости) s r, %
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), s R, %
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
Спектрофотометр или фотоколориметр, позволяющий измерять оптическую плотность при длине волны l = 520 нм
Кюветы с толщиной поглощающего слоя 20 мм
Весы лабораторные 2 класса точности, ГОСТ 24104
ГСО с аттестованным содержанием ионов свинца
Воронки делительные ВД-3-1000 ХС, ГОСТ 25336.
Колбы конические Кн-1-1000-29/32, ГОСТ 25336.
Холодильники ХШ-1-200-29/32 ХС, ГОСТ 25336.
Бумага универсальная индикаторная, ТУ-6-09-1181.
Баня водяная, ТУ 46-22-606-75.
Бутыли из стекла или полиэтилена с притертыми или винтовыми пробками вместимостью 500 — 1000 см 3 для отбора и хранения проб и реактивов.
Феноловый красный, ГОСТ 4599.
Четыреххлористый углерод, ГОСТ 20288 (продажный реактив перегоняют, собирая фракцию, кипящую при 76 °С).
Натрий лимоннокислый, ТУ 6-09-2248-78.
Калий железистосинеродистый трехводный, ГОСТ 4207. ( * )
Аскорбиновая кислота, ГОСТ 4815. ( * )
Вес реактивы должны быть квалификации х.ч. или ч.д.а.
4.1 . При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007 .
4.2 . Электробезопасность при работе с электроустановками по ГОСТ 12.1.019 .
4.3 . Организация обучения работающих безопасности труда по ГОСТ 12.0.004 .
4.4 . Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009 .
Выполнение измерений может производить химик аналитик, владеющий техникой экстракционно-фотометрического анализа и изучивший инструкцию по эксплуатации спектрофотометра или фотоколориметра.
Измерения проводятся в следующих условиях:
температура окружающего воздуха (20 ± 5) °С;
атмосферное давление (84,0 — 106,7) кПа (630 — 800 мм. рт. ст);
относительная влажность (80 ± 5) %;
напряжение сети (220 ± 10) В;
частота переменного тока (50 ± 1) Гц.
Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб». ( * )
7.1 . Бутыли для отбора и хранения проб воды обезжиривают раствором CMC, промывают водопроводной водой, обрабатывают хромовой смесью, тщательно промывают водопроводной, затем 3 — 4 раза дистиллированной водой.
7.2 . Пробы воды (объем не менее 1000 см 3 ) отбирают в полиэтиленовые бутыли, предварительно ополоснутые отбираемой водой.
7.3 . Пробы анализируют не позднее, чем через 2 часа после отбора или консервируют следующим образом: к пробе добавляют 3 см 3 концентрированной HN О3 на 1 дм 3 воды, или 3 см 3 ледяной уксусной кислоты на 1 дм 3 воды. Срок хранения законсервированной пробы — 1 месяц.
Если требуется определить свинец в растворенной форме, пробу фильтруют через бумажный фильтр «синяя лента» до консервирования ( * ) .
7.4 . Проба воды не должна подвергаться воздействию прямого солнечного света. Для доставки в лабораторию сосуды с пробами упаковывают в тару, обеспечивающую сохранность и предохраняющую от резких перепадов температуры. При отборе проб составляют сопроводительный документ по утвержденной форме, в котором указывают:
цель анализа, предполагаемые загрязнители;
должность, фамилия отбирающего пробу, дата.
Подготовку прибора к работе и оптимизацию условий измерения производят в соответствии с рабочей инструкцией по эксплуатации прибора. Прибор должен быть поверен.
Дважды перегоняют воду в приборе из стекла, не содержащего свинца, и проверяют ее на отсутствие свинца раствором дитизона.
300 — 500 см 3 воды встряхивают с 20 см 3 раствора дитизона (по п. 8.2.9 ). Процедуру повторяют до тех пор пока цвет дитизона не будет оставаться зеленым. Избыток дитизона удаляют встряхиванием бидистиллированной воды с четыреххлористым углеродом. Дитизон считают полностью удаленным, если после очередного встряхивания слой растворителя остается бесцветным.
Для приготовления всех реактивов и растворов используют эту воду.
0 ,1 г индикатора растворяют в 100 см 3 20 %-ного раствора спирта-ректификата.
100 г натрия лимоннокислого растворяют в небольшом количестве воды, помещают в мерную колбу на 500 см 3 и доводят до метки бидистиллированной водой.
Очищают от микроэлементов экстракцией в делительной воронке раствором дитизона (по п. 8.2.9.) порциями по 5 — 10 см 3 до прекращения окраски дитизона. Продолжительность каждой экстракции 1 мин. После этого из раствора извлекают остатки дитизона экстракцией четыреххлористым углеродом порциями по 10 см 3 до полного удаления дитизона, т.е. пока очередная порция четыреххлористого углерода не станет бесцветной. Продолжительность каждой экстракции 1 мин.
500 см 3 соляной кислоты осторожно приливают к 500 см 3 бидистиллированной воды.
8.2.4 . Калий железистосинеродистый ( * ) , водный раствор.
Навеску (1 г) калия железистосинеродистого ( * ) помещают в мерную колбу на 50 см 3 , растворяют в небольшом количестве воды, перемешивают и доводят до метки бидистиллированной водой.
Навеску (10 г) гидроксиламина солянокислого помещают в мерную колбу на 500 см 3 , растворяют в небольшом количестве воды, перемешивают и доводят до метки бидистиллированной водой.
Раствор очищают экстракцией раствором дитизона (по п. 8.2.9.) порциями по 5 — 10 см 3 до прекращения изменения окраски дитизона. Продолжительность каждой экстракции 1 мин. После этого из раствора извлекают остатки дитизона экстракцией четыреххлористым углеродом порциями по 10 см 3 до полного удаления дитизона, т.е. пока очередная порция четыреххлористого углерода не станет бесцветной. Продолжительность каждой экстракции 1 мин.
8.2.6 . Приготовление основного градуировочного раствор свинца.
Раствор готовят из ГСО с аттестованным содержанием свинца в соответствии с прилагаемой к образцу инструкцией.
Раствор устойчив в течение года и хранится в склянке с притертой пробкой при комнатной температуре.
1 см 3 раствора должен содержать 0,1 мг свинца.
8.2.7 . Приготовление рабочего градуировочного раствора свинца.
Рабочий раствор готовят в день проведения анализа разбавлением основного раствора в 100 раз бидистиллированной водой.
1 см 3 раствора должен содержать 0,001 мг свинца.
8.2.8 . Приготовление основного раствора дитизона.
0 ,05 г дитизона очищенного (см. приложение) растворяют в 500 см 3 четыреххлористого углерода. Раствор хранят в темной склянке при 3 — 5 °С в течение нескольких месяцев.
8.2.9 . Приготовление рабочего раствора дитизона.
25 см 3 основного раствора дитизона разбавляют четыреххлористым углеродом и объём доводят до 250 см 3 . Измеряют оптическую плотность полученного раствора ( l = 520 нм, кювета 20 мм) против четыреххлористого углерода. Прибавляя в колбу по каплям раствор дитизона (по п. 8.2.8) или четыреххлористого углерода, устанавливают оптическую плотность, равную 0,350. Раствор готовят в день определения и хранят в темной склянке ( * ) .
8.2.10 . Приготовление раствора аммиака, 17 %-ный раствор.
Раствор получают путем изопиестической дистилляции концентрированного раствора аммиака. Для этого на дно эксикатора наливают 1 дм 3 концентрированного аммиака, а на вкладыш ставят выпарительную чашку с 500 см 3 бидистиллированной воды. Через двое суток аммиак, полученный в чашке, будет иметь концентрацию примерно 17 %.
Для построения градуировочного графика необходимо приготовить образцы для градуировки определяемого компонента с концентрациями 0,002 — 0,03 мг/дм 3 . Условия анализа, его проведение должны соответствовать описанным в пунктах 6 и 9.
Состав и количество образцов для градуировки для построения градуировочного графика приведены в таблице 2. Погрешность, обусловленная процедурой приготовления образцов для градуировки, не превышает 2,8 %.
Состав и количество образцов для градуировки при анализе свинца
Массовая концентрация свинца в градуировочных растворах, мг/дм 3
Аликвотная часть рабочего раствора с концентрацией 0,001 мг/см 3 , помещенного в мерную колбу на 500 см 3
В качестве холостой пробы используют бидистиллированную воду (п. 8.2), которую проводят через весь ход анализа ( * ) .
Растворы готовят в мерных колбах вместимостью 500 см 3 . Полученные растворы переносят в конические колбы со шлифом емкостью 1 дм 3 , подкисляют 10 см 3 раствором соляной кислоты (по п. 8.2.3), прибавляют по 0,5 г персульфата аммония, вставляют в горла колб пробки-холодильники. Кипятят в течение 20 минут. Далее поступают как указано в п. 9.
Анализ образцов для градуировки проводят в порядке возрастания их концентрации. Для построения градуировочного графика каждую искусственную смесь необходимо фотометрировать 3 раза с целью исключения случайных результатов и усреднения данных. Из оптической плотности каждого градуировочного раствора вычитают оптическую плотность холостой пробы ( * ) .
Строят градуировочный график в координатах оптическая плотность — концентрация в мг/дм 3 .
Контроль стабильности градуировочной характеристики проводят не реже одного раза в месяц или при смене партии реактивов. Средствами контроля являются вновь приготовленные образцы для градуировки (не менее 3 образцов из приведенных в таблице 2).
Градуировочную характеристику считают стабильной при выполнении для каждого образца для градуировки следующего условия:
где Х — результат контрольного измерения массовой концентрации ионов свинца в образце для градуировки;
С — аттестованное значение массовой концентрации ионов свинца в образце для градуировки;
s Rл— среднеквадратическое отклонение внутрилабораторной прецизионности, установленное при реализации методики в лаборатории.
Примечание. Допустимо среднеквадратическое отклонение внутрилабораторной прецизионности при внедрении методики в лаборатории устанавливать на основе выражения: s Rл = 0,84 s R, с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Значения s R приведены в таблице 1.
Если условие стабильности градуировочной характеристики не выполняется только для одного образца для градуировки, необходимо выполнить повторное измерение этого образца с целью исключения результата, содержащего грубую погрешность.
Если градуировочная характеристика нестабильна, выясняют причины и повторяют контроль с использованием других образцов для градуировки, предусмотренных методикой. При повторном обнаружении нестабильности градуировочной характеристики строят новый градуировочный график.
0 ,5 дм 3 исследуемой воды, предварительно подкисленной (20 см 3 соляной кислоты по п. 8.2.3 на 1 дм 3 пробы), помещают в колбу на 1 дм 3 , прибавляют 0,5 г персульфата аммония и, вставив в горло колбы пробку-холодильник, кипятят на медленном огне в течение 20 мин., после чего пробу охлаждают под струей воды.
К охлажденной пробе приливают
15 см 3 аммиака (по п. 8.2.10), затем по каплям доводят кислотность среды до рН = 2 (контроль по универсальной индикаторной бумаге). Пробу переносят в делительную воронку емкостью 1 дм 3 и экстрагируют 10 см 3 раствора дитизона (по п. 8.2.9 ) в течение 2 минут. После расслоения водной и органической фаз экстракт сливают и отбрасывают. Повторяют экстракцию до тех пор, пока слой органического растворителя не перестанет изменять окраску.
К очищенной пробе прибавляют 5 — 6 капель раствора фенолового красного (по п. 8.2.1) и, приливая по каплям аммиак (по п. 8.2.10), нейтрализуют пробу до оранжевой окраски (рН = 6,8 — 7,3). Затем приливают 5 см 3 раствора калия железистосинеродистого ( * ) (по п. 8.2.4), 5 см 3 раствора солянокислого гидроксиламина (по п. 8.2.5), 5 см 3 раствора лимоннокислого натрия (по п. 8.2.2) и встряхивают содержимое воронки в течение 30 сек. Затем прибавляют по каплям аммиак (по п. 8.2.10) до появления малиновой окраски и избыток
5 капель (рН = 8,0 — 8,5). Приливают 10 см 3 раствора дитизона (по п. 8.2.9) и экстрагируют свинец в течение 2 минут. После расслоения сливают окрашенный слой органического растворителя в кювету, фильтруя его через воронку с небольшим слоем ваты. Измеряют оптическую плотность раствора при длине волны l = 520 нм в кювете с толщиной слоя 20 мм против четыреххлористого углерода. Вычитают оптическую плотность холостой пробы (см. п. 8.3) ( * ) . Содержание свинца находят по градуировочному графику.
Содержание ионов свинца (мг/дм 3 ) рассчитывают по формуле
где С — концентрация свинца, найденная по градуировочному графику, мг/дм 3 ;
500 — объем, до которого была разбавлена проба, см 3 ;
V — объем, взятый для анализа, см 3 .
За результат анализа Хсрпринимают среднее арифметическое значение двух параллельных определений Х1и Х2
для которых выполняется следующее условие:
где r — предел повторяемости, значения которого приведены в таблице 3.
Значения предела повторяемости вероятности Р = 0,95
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных определений), r, %
При невыполнении условия (1) могут быть использованы методы проверки приемлемости результатов параллельных определений и установления окончательного результата согласно раздела 5 ГОСТ Р ИСО 5725-3.
Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 4.
Значения предела воспроизводимости при вероятности Р = 0,95
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R, %
При превышении предела воспроизводимости могут быть использованы методы оценки приемлемости результатов анализа согласно раздела 5 ГОСТ Р ИСО 5725-6.
11.1 . Результат анализа X ср в документах, предусматривающих его использование, может быть представлен в виде: Хср± D , Р = 0,95,
где D — показатель точности методики.
Значение D рассчитывают по формуле: D = 0,01 × d × Хср . Значение d приведено в таблице 1.
Допустимо результат анализа в документах, выдаваемых лабораторией, представлять в виде: Хср± D л , Р = 0,95, при условии D л D ,
где Хср— результат анализа, полученный в соответствии с прописью методики;
± D л — значение характеристики погрешности результатов анализа, установленное при реализации методики в лаборатории и обеспечиваемое контролем стабильности результатов анализа.
Примечание. При представлении результата анализа в документах, выдаваемых лабораторией, указывают:
— количество результатов параллельных определений, использованных для расчета результата анализа;
— способ определения результата анализа (среднее арифметическое значение или медиана результатов параллельных определений).
11.2 . В том случае, если массовая концентрация ионов свинца в анализируемой пробе превышает верхнюю границу диапазона, то допускается разбавление пробы таким образом, чтобы массовая концентрация ионов свинца соответствовала регламентированному диапазону.
Результат анализа Хсрв документах, предусматривающих его использование, может быть представлен в виде: Хср± D ’, Р = 0,95,
где ± D ’ — значение характеристики погрешности результатов анализа, откорректированное на величину погрешности взятия аликвоты.
11.3 . Если массовая концентрация ионов свинца в анализируемой пробе ниже минимально определяемой по методике концентрации, то допускается концентрирование. В этом случае одновременно с анализируемой пробой ведут анализ аттестованного (стандартного) раствора с содержанием ионов свинца, соответствующим содержанию их в исходной рабочей пробе. Результат анализа исходной рабочей пробы признают удовлетворительным, если выполняется следующее условие:
где X — результат контрольного измерения массовой концентрации ионов свинца в образце для контроля (стандартном растворе);
С — аттестованное значение массовой концентрации ионов свинца в образце для контроля (стандартном растворе);
К — норматив оперативного контроля процедуры анализа.
D ” — значение характеристики погрешности результатов анализа, откорректированное на величину концентрирования пробы.
Контроль качества результатов анализа при реализации методики в лаборатории предусматривает:
— оперативный контроль процедуры анализа (на основе оценки погрешности при реализации отдельно взятой контрольной процедуры);
— контроль стабильности результатов анализа (на основе контроля стабильности среднеквадратического отклонения повторяемости, среднеквадратического отклонения внутрилабораторной прецизионности, погрешности).
12.1 . Алгоритм оперативного контроля процедуры анализа с использованием метода добавок
Оперативный контроль процедуры анализа проводят путем сравнения результата отдельно взятой контрольной процедуры Ккс нормативом контроля К.
Результат контрольной процедуры Кк рассчитывают по формуле:
где Х ¢ ср — результат анализа массовой концентрации ионов свинца в пробе с известной добавкой — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 10;
Хср — результат анализа массовой концентрации ионов свинца в исходной пробе — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 10.
Норматив контроля К рассчитывают по формуле
где 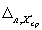 ,
, 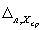 — значения характеристики погрешности результатов анализа, установленные в лаборатории при реализации методики, соответствующие массовой концентрации ионов свинца в пробе с известной добавкой и в исходной пробе соответственно.
— значения характеристики погрешности результатов анализа, установленные в лаборатории при реализации методики, соответствующие массовой концентрации ионов свинца в пробе с известной добавкой и в исходной пробе соответственно.
Примечание. Допустимо характеристику погрешности результатов анализа при внедрении методики в лаборатории устанавливать на основе выражения: D л = 0,84 · D , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Процедуру анализа признают удовлетворительной, при выполнении условия:
При невыполнении условия (2) контрольную процедуру повторяют. При повторном невыполнении условия (2) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры по их устранению.
12.2 . Алгоритм оперативного контроля процедуры анализа с применением образцов для контроля
Оперативный контроль процедуры анализа проводят путем сравнения результата отдельно взятой контрольной процедуры Ккс нормативом контроля К.
Результат контрольной процедуры Кк рассчитывают по формуле:
где C ср — результат анализа массовой концентрации ионов свинца в образце для контроля — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 10;
С — аттестованное значение образца для контроля.
Норматив контроля К рассчитывают по формуле:
где ± D л — характеристика погрешности результатов анализа, соответствующая аттестованному значению образца для контроля.
Примечание. Допустимо характеристику погрешности результатов анализа при внедрении методики в лаборатории устанавливать на основе выражения: D л = 0,84 · D , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Процедуру анализа признают удовлетворительной, при выполнении условия:
При невыполнении условия (3) контрольную процедуру повторяют. При повторном невыполнении условия (3) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры по их устранению.
Периодичность оперативного контроля процедуры анализа, а также реализуемые процедуры контроля стабильности результатов анализа регламентируют в Руководстве по качеству лаборатории.
В 100 см 3 четыреххлористого углерода растворяют 1,0 г дитизона. Раствор переносят в делительную воронку на 500 см 3 , приливают 100 см 3 раствора аммиака (1 см 3 концентрированного аммиака, разбавленного до 100 см 3 бидистиллированной водой) и 5 см 3 5 %-ного раствора аскорбиновой кислоты. Содержимое воронки встряхивают в течение 2 мин. После расслоения жидкости органический слой сливают в чистую делительную воронку. Водный раствор дитизона фильтруют в колбу на 1 дм 3 через бумажный фильтр, предварительно промытый раствором соляной кислоты (1:20) и бидистиллированной водой. К органическому раствору дитизона приливают новую порцию раствора аммиака и раствора аскорбиновой кислоты, и содержимое воронки встряхивают в течение 2 мин. Операцию очистки дитизона повторяют 5 — 6 раз до тех пор, пока водно-аммиачный раствор не будет окрашиваться в оранжевый цвет. Все порции водного раствора дитизона фильтруют в колбу на 1 дм 3 . Приливают соляную кислоту (1:1) до выпадения дитизона в осадок. При этом водный слой приобретает бледно-зеленоватый цвет. Осадок фильтруют через бумажный фильтр, промывают 3 раза 1 %-ным раствором аскорбиновой кислоты и сушат на воздухе. Дитизон хранят в темном месте.
источник
ПНД Ф 14.1:2:3:4.239-2007. Количественный химический анализ вод. Методика измерений массовой концентрации свинца в питьевых, поверхностных, подземных пресных и сточных водах хроматным фотометрическим методом с дифенилкарбазидом
| Наименование документа: | ПНД Ф 14.1:2:3:4.239-2007 |
| Тип документа: | ПНД Ф |
| Статус документа: | Действует |
| Название: | Количественный химический анализ вод. Методика измерений массовой концентрации свинца в питьевых, поверхностных, подземных пресных и сточных водах хроматным фотометрическим методом с дифенилкарбазидом |
| Область применения: | Документ устанавливает методику измерений свинца в питьевых, поверхностных, подземных пресных и сточных водах хроматным фотометрическим методом с дифенилкарбазидом. Диапазон измерений от 0,04 до 2 мг/дм3 |
| Краткое содержание: | |
| Дата добавления в базу: | 01.09.2013 |
| Дата актуализации: | 01.12.2013 |
| Доступно сейчас для просмотра: | 100% текста. Полная версия документа. |
| Организации: |