ПНД Ф 14.1:2.100-97
Количественный химический анализ вод. Методика выполнения измерений химического потребления кислорода в пробах природных и очищенных сточных вод титриметрическим методом
Купить ПНД Ф 14.1:2.100-97 — бумажный документ с голограммой и синими печатями. подробнее
Распространяем нормативную документацию с 1999 года. Пробиваем чеки, платим налоги, принимаем к оплате все законные формы платежей без дополнительных процентов. Наши клиенты защищены Законом. ООО «ЦНТИ Нормоконтроль».
Наши цены ниже, чем в других местах, потому что мы работаем напрямую с поставщиками документов.
- Срочная курьерская доставка (1-3 дня)
- Курьерская доставка (7 дней)
- Самовывоз из московского офиса
- Почта РФ
Документ устанавливает методику количественного химического анализа проб природных и очищенных сточных вод для определения в них величины химического потребления кислорода (ХПК) при содержании органических веществ, эквивалентном потреблению молекулярного кислорода в диапазоне от 4,0 до 80,0 мг/дм3 титриметрическим методом без концентрации пробы.
Методика допущена для целей государственного экологического контроля
3. Приписанные характеристики погрешности измерений и ее составляющих
4. Средства измерений, вспомогательные устройства, материалы, реактивы
5. Требования безопасности
6. Требования к квалификации операторов
9. Подготовка к выполнению измерений
10. Устранение мешающих влияний
12. Обработка результатов измерений
13. Оформление результатов анализа
14. Контроль качества результатов анализа при реализации методики в лаборатории
×
| Дата введения: | 01.12.2016 |
|---|---|
| Добавлен в базу: | 01.09.2013 |
| Заверение срока действия: | 01.12.2016 |
| Актуализация: | 01.01.2019 |
Чтобы бесплатно скачать этот документ в формате PDF, поддержите наш сайт и нажмите кнопку:
ГОСУДАРСТВЕННЫЙ КОМИТЕТ РОССИЙСКОЙ
ФЕДЕРАЦИИ ПО ОХРАНЕ ОКРУЖАЮЩЕЙ СРЕДЫ
Государственного комитета РФ
по охране окружающей среды
_____________ А.А. Соловьянов
КОЛИЧЕСТВЕННЫЙ ХИМИЧЕСКИЙ АНАЛИЗ ВОД
МЕТОДИКА ВЫПОЛНЕНИЯ ИЗМЕРЕНИЙ
ХИМИЧЕСКОГО ПОТРЕБЛЕНИЯ КИСЛОРОДА
В ПРОБАХ ПРИРОДНЫХ И ОЧИЩЕННЫХ СТОЧНЫХ ВОД
ТИТРИМЕТРИЧЕСКИМ МЕТОДОМ
Методика допущена для целей государственного экологического контроля
МОСКВА 1997 г.
(издание 2004 г.)
Настоящий документ устанавливает методику количественного химического анализа проб природных и очищенных сточных вод для определения в них величины химического потребления кислорода (ХПК) при содержании органических веществ, эквивалентном потреблению молекулярного кислорода в диапазоне от 4,0 до 80,0 мг/дм 3 титриметрическим методом без концентрирования пробы.
При величине ХПК > 50 мг/дм 3 определение следует проводить при соответствующем разбавлении пробы дистиллированной водой.
Определению мешают хлориды, сульфиды, соединения железа(II), нитриты и другие неорганические вещества, способные окисляться бихроматом в кислой среде.
Мешающие влияния устраняют в соответствии с п. 10.
Титриметрический метод определения ХПК основан на окислении органических веществ избытком бихромата калия в растворе серной кислоты при нагревании в присутствии катализатора — сульфата серебра. Остаток бихромата калия находят титрованием раствором соли Мора и по разности определяют количество K2Cr2O7, израсходованное на окисление органических веществ.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведённых в таблице 1.
Диапазон измерений, значения показателей точности, повторяемости, воспроизводимости
Диапазон измерений величины ХПК, мг/дм 3
Показатель точности (границы относительной погрешности при вероятности Р = 0,95),
±d, %
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости),
sr, %
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости),
sR, %
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
Весы лабораторные общего назначения с наибольшим пределом
взвешивания 200 г и ценой наименьшего деления 0,1 мг любого типа
Весы лабораторные общего назначения с наибольшим пределом
взвешивания 200 г и ценой наименьшего деления 10 мг любого типа
СО с аттестованным содержанием ХПК с погрешностью не более 1 % при Р = 0,95
Цилиндры мерные или мензурки
4.2. Вспомогательные устройства
Плитки электрические с закрытой спиралью и регулируемой
мощностью нагрева
Шкаф сушильный лабораторный с температурой нагрева до 130 °С
Стаканчики для взвешивания (бюксы)
Установки для определения ХПК в составе:
Колба К-1-250-29/32 ТС или колба Гр-250-29/32
Обратный холодильник ХПТ-2-400-29/32 ХС
Прибор вакуумного фильтрования ПВФ-35 или ПВФ-47
Средства измерений должны быть поверены в установленные сроки.
Допускается использование других, в том числе импортных, средств измерений и вспомогательных устройств с характеристиками не хуже, чем у приведенных в п.п. 4.1 и 4.2.
Бихромат калия (калий двухромовокислый)
N-фенилантраниловая кислота или
Бумага индикаторная универсальная
Фильтры мембранные Владипор типа МФАС-МА или МФАС-ОС-2 (0,45 мкм)
или фильтры бумажные обеззоленные «синяя лента»
Все реактивы, используемые для анализа, должны быть квалификации ч.д.а. или х.ч.
Допускается использование реактивов, изготовленных по другой нормативно-технической документации, в том числе импортных, с квалификацией не ниже ч.д.а.
5.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
5.2. Электробезопасность при работе с электроустановками обеспечивается по ГОСТ 12.1.019.
5.3. Организация обучения работающих безопасности труда проводится по ГОСТ 12.0.004.
5.4. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Выполнение измерений может производить химик-аналитик, владеющий техникой титриметрического метода анализа.
При выполнении измерений в лаборатории должны быть соблюдены следующие условия:
· температура окружающего воздуха (22 ± 6) °С;
· атмосферное давление (84 — 106) кПа;
· относительная влажность не более 80 % при температуре 25 °С;
· частота переменного тока (50 ± 1) Гц;
· напряжение в сети (220 ± 22) В.
8.1. Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб».
8.2. Посуду, предназначенную для отбора и хранения проб, моют хромовой смесью, затем тщательно (не менее 10 раз) промывают водопроводной и ополаскивают дистиллированной водой.
8.3. Пробы воды отбирают в стеклянную посуду с пробками, не загрязняющими пробу органическими соединениями.
В зависимости от целей анализа определение ХПК можно проводить в нефильтрованной или фильтрованной пробе. В последнем случае пробу предварительно фильтруют через мембранный фильтр 0,45 мкм, очищенный двухкратным кипячением в дистиллированной воде. Допустимо использование бумажных фильтров «синяя лента», промытых дистиллированной водой. При фильтровании через любой фильтр первые порции фильтрата отбрасывают.
Объем отбираемой пробы должен быть не менее 100 см 3 .
8.4. Определение ХПК, особенно в загрязненных водах, следует проводить как можно скорее после отбора пробы. Допускается хранение пробы при температуре не выше 4 °С не более суток при консервации добавлением раствора серной кислоты (1:2) из расчета 2 см 3 на каждые 100 см 3 пробы воды.
8.5. При отборе проб составляется сопроводительный документ по утвержденной форме, в котором указывается:
— цель анализа, предполагаемые загрязнители;
— должность, фамилия отбирающего пробу, дата.
9.1. Приготовление растворов и реактивов
9.1.1. Раствор бихромата калия с концентрацией 0,25 моль/дм 3 эквивалента.
6,129 г бихромата калия, предварительно высушенного в течение 2 ч при 105 °С, количественно переносят его в мерную колбу вместимостью 500 см 3 , растворяют в дистиллированной воде, доводят до метки и перемешивают. Раствор устойчив при хранении в плотно закрытой темной склянке в течение 6 мес.
9.1.2. Раствор бихромата калия с концентрацией 0,025 моль/дм 3 эквивалента.
50 см 3 раствора бихромата калия с концентрацией 0,25 моль/дм 3 эквивалента помещают в мерную колбу вместимостью 500 см 3 и доводят объем раствора до метки дистиллированной водой. Хранят в склянке с притертой пробкой в темном месте не более 6 мес.
9.1.3. Раствор соли Мора с концентрацией 0,25 моль/дм 3 эквивалента.
49,0 г соли Мора переносят в мерную колбу вместимостью 500 см 3 , растворяют в дистиллированной воде, осторожно добавляют 10 см 3 концентрированной серной кислоты и после охлаждения доводят объем раствора до метки дистиллированной водой. Хранят в плотно закрытой посуде не более 6 мес.
9.1.4. Раствор соли Мора с концентрацией 0,025 моль/дм 3 эквивалента.
50 см 3 раствора соли Мора с концентрацией 0,25 моль/дм 3 эквивалента помещают в мерную колбу вместимостью 500 см 3 и доводят объем раствора до метки дистиллированной водой. Хранят в плотно закрытой посуде не более 3 мес.
Точную концентрацию раствора устанавливают ежедневно или перед серией определений в соответствии с п. 10.2.
В качестве индикатора используют раствор N-фенилантраниловой кислоты или ферроина (комплекс сульфата железа(II) с 1,10-фенантролином).
Для приготовления раствора N-фенилантраниловой кислоты 0,25 г реактива растворяют в 12 см 3 раствора гидрооксида натрия (для ускорения процесса раствор можно слегка подогреть) и разбавляют дистиллированной водой до 250 см 3 .
Для приготовления раствора ферроина 2,43 г индикатора растворяют в 100 см 3 дистиллированной воды.
При приготовлении раствора ферроина на основе 1,10-фенантролина растворяют 0,980 г соли Мора (NH4)2Fe(SO4)2 · 6H2O в 100 см 3 дистиллированной воды, добавляют 2,085 г 1,10-фенантролина моногидрата или 2,93 г сульфата и перемешивают до растворения последнего.
Раствор индикатора хранят в плотно закрытой склянке из темного стекла не более 3 мес.
9.1.6. Раствор гидроксида натрия, 0,4 %.
0,4 г NaOH растворяют в 100 см 3 дистиллированной воды. Раствор устойчив при хранении в плотно закрытой полиэтиленовой посуде не более 2 мес.
9.1.7. Раствор сульфата серебра.
5,0 г Ag2SO4 растворяют в 1 дм 3 концентрированной серной кислоты. Раствор устойчив в склянке из темного стекла в течение 6 мес.
9.2. Установление точной концентрации раствора соли Мора
Пипеткой вместимостью 10 см 3 отбирают 10 см 3 раствора бихромата калия с концентрацией 0,025 моль/дм 3 эквивалента (п. 9.1.2), переносят в коническую колбу, добавляют 180 см 3 дистиллированной воды и 20 см 3 концентрированной серной кислоты. После охлаждения добавляют в пробу 3 — 4 капли индикатора ферроина или 10 капель раствора N-фенилантраниловой кислоты и титруют раствором соли Мора с концентрацией 0,025 моль/дм 3 эквивалента (п. 9.1.4) до перехода окраски из синевато-зеленой в красно-коричневую при использовании в качестве индикатора ферроина и из красно-фиолетовой в синевато-зеленую при использовании N-фенилантраниловой кислоты.
Титрование повторяют и при отсутствии расхождения в объемах титранта более 0,05 см 3 за результат принимают среднее значение. В противном случае повторяют титрование до получения результатов, отличающихся не более, чем на 0,05 см 3 .
Точную концентрацию раствора соли Мора находят по формуле:
где См — концентрация раствора соли Мора, моль/дм 3 эквивалента;
Сб — концентрация раствора бихромата калия, моль/дм 3 эквивалента;
Vб — объем раствора бихромата калия, взятый для титрования, см 3 ;
Vм — объем раствора соли Мора, пошедший на титрование см 3 .
Мешающее влияние хлоридов при концентрациях менее 300 мг/дм 3 устраняется за счет присутствия в пробе катализатора (сульфата серебра). При больших содержаниях хлоридов к пробе добавляют сульфат ртути (II) из расчета 100 мг на 10 мг хлоридов.
Мешающее влияние сульфидов и соединений железа (II) устраняют предварительной продувкой пробы воды воздухом, если она не содержит летучих органических соединений, или учитывают при расчете ХПК. В последнем случае определяют их концентрации и пересчитывают на величины ХПК, исходя из того, что 1 мг H2S и 1 мг Fe 2+ эквивалентны соответственно 0,47 и 0,14 мг O2. Таким же образом учитывают влияние нитритов (1 мг NО2 эквивалентен 0,35 мг O2).
11.1. Выполнение измерений в водах с низкой концентрацией хлоридов
Если концентрация хлоридов в пробе анализируемой воды составляет менее 300 мг/дм 3 , в колбу со шлифом установки для определения ХПК вносят с помощью пипетки 20 см 3 воды (или аликвоту, доведенную дистиллированной водой до 20 см 3 ), добавляют 10,0 см 3 раствора бихромата калия с концентрацией 0,025 моль/дм 3 эквивалента (п. 9.1.2) и 30 см 3 раствора сульфата серебра в концентрированной серной кислоте. Для равномерного кипения в колбу бросают 2 — 3 капилляра, присоединяют к ней обратный холодильник и кипятят содержимое на песчаной бане в течение 2 ч.
После охлаждения установки промывают холодильник дистиллированной водой (около 50 см 3 ), отсоединяют его, добавляют в колбу, обмывая ее стенки, еще 50 см 3 дистиллированной воды, вновь охлаждают, переносят пробу в коническую колбу, дважды споласкивая колбу, где кипятилась проба, дистиллированной водой (по 20 — 30 см 3 ). Добавляют 3 — 4 капли раствора ферроина (или 10 капель раствора фенилантраниловой кислоты) и титруют избыток непрореагировавшего бихромата калия раствором соли Мора (п. 9.1.4) до перехода окраски индикатора из синевато-зеленой в красно-коричневую при использовании в качестве индикатора ферроина и из красно-фиолетовой в синевато-зеленую при использовании N-фенилантраниловой кислоты.
Аналогичным образом проводят холостой опыт с 20 см 3 дистиллированной воды.
11.2. Выполнение измерений в водах с высокой концентрацией хлоридов
Если концентрация хлоридов в воде превышает 300 мг/дм 3 , к отобранной для анализа пробе (20 см 3 или меньшей аликвоте, доведенной до 20 см 3 дистиллированной водой) добавляют сульфат ртути из расчета 100 мг на каждые 10 мг содержащихся в пробе хлоридов и тщательно перемешивают. Далее выполняют определение, как описано в п. 11.1. Наличие небольшого количества осадка, образовавшегося после добавления сульфата ртути, не мешает определению.
12.1. Величину ХПК (бихроматной окисляемости) анализируемой пробы воды X находят по формуле:
где Vмх — объем раствора соли Мора, израсходованный на титрование в холостом опыте, см 3 ;
Vм — объем раствора соли Мора, израсходованный на титрование в пробы воды, см 3 ;
См — концентрация раствора соли Мора, моль/дм 3 эквивалента;
V — объем пробы воды, взятый для определения, см 3 ;
8,0 — масса миллиграмм-эквивалента кислорода, мг.
Если величина ХПК в анализируемой пробе превышает верхнюю границу диапазона (80 мг/дм 3 ), разбавляют пробу с таким расчетом, чтобы величина ХПК входила в регламентированный диапазон, и выполняют определение в соответствии с п. 11.2.
В этом случае величину ХПК в анализируемой пробе воды X находят по формуле:
где ХV — величина ХПК в разбавленной пробе воды, мг/дм 3 ;
VV— объем пробы воды после разбавления, см 3 ;
v — объем аликвоты пробы воды, взятой для разбавления, см 3 .
12.2. Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 2.
Значения предела воспроизводимости при вероятности Р = 0,95
Диапазон измерений величины ХПК, мг/дм 3
Предел воспроизводимости (относительное значение допускаемого расхождения
между двумя результатами измерений, полученными в разных лабораториях), R, %
При превышении предела воспроизводимости могут быть использованы методы оценки приемлемости результатов анализа согласно раздела 5 ГОСТ Р ИСО 5725-6.
Результат анализа X в документах, предусматривающих его использование, может быть представлен в виде:
где D — показатель точности методики.
Значение D рассчитывают по формуле:
Значение d приведено в таблице 1.
Если проводилось разбавление пробы воды из-за превышения величины ХПК верхней границы диапазона, значение d выбирают из таблицы 1 для величины ХПК в разбавленной пробе воды ХV.
Допустимо результат анализа в документах, выдаваемых лабораторией, представлять в виде:
источник
Фотометрический метод определения массовой концентрации ионов аммония основан на взаимодействии NH 4 + — иoнов с тетраиодомеркуратом калия в щелочной среде K 2 HgI 4 + КОН (реактив Несслера) с образованием коричневой, нерастворимой в воде соли основания Миллона [ Hg 2 N] ∙ H 2 O , переходящей в коллоидную форму при малых содержаниях NH 4 + — иoнов . Светопоглощение раствора измеряют при λ = 425 нм в кюветах с длиной поглощающего слоя 1 или 5 см. Интенсивность окраски прямо пропорциональна концентрации NH 4 + — ионов в растворе пробы.
Настоящая методика обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведенных в таблице 1.
Диапазон измерений, значения показателей точности, повторяемости и воспроизводимости
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ± δ, %
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости) s r, %
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости) s R , %
Значения показателя точности методики используют при:
— оформлении результатов анализа, выдаваемых лабораторией;
— оценке деятельности лабораторий на качество проведения испытаний;
— оценке возможности использования результатов анализа при реализации методики в конкретной лаборатории.
Спектрофотометр или фотоэлектроколориметр, измеряющий светопоглощение при λ= 425 нм.
Кюветы с длиной поглощающего слоя 10 и 50 мм.
Весы лабораторные 2 класса точности ГОСТ 24104.
Колбы мерные, наливные 2-50-2
Колбы плоскодонные Кн-2-500-18 ТСХ ГОСТ 25336.
Стаканчики для взвешивания СВ ГОСТ 25336.
Аппарат для обыкновенной перегонки или с водяным паром (аппарат Парнаса-Вагнера).
Сушильный шкаф электрический ОСТ 16.0.801.397.
Фильтры обеззоленные ТУ 6-09-1678.
Бумага индикаторная, универсальная ТУ-6-09-1181.
Воронки стеклянные для фильтрования ГОСТ 25336.
Бутыли из стекла или полиэтилена с притертыми или винтовыми пробками вместимостью 500 — 1000 см 3 для отбора и хранения проб и реактивов.
Стандартный образец с аттестованным содержанием ионов аммония или аммоний хлористый, ГОСТ 3773.
Реактив Несслера, ТУ 6-09-2089.
Калий фосфорнокислый однозамещенный, ГОСТ 4198.
Калий фосфорнокислый двузамещенный, ГОСТ 2493.
Калия гидроокись, ТУ 6-09-50-2322.
Натрий мышьяковистокислый (метаарсенит), ТУ 6-09-2791.
Натрий серноватистокислый (тиосульфат), СТ СЭВ 223.
Калий-натрий виннокислый 4-х водный (сегнетова соль), ГОСТ 5845.
Этилендиамин-N,N,N’N’-тетрауксусной кислоты динатриевая соль (Трилон Б) ГОСТ 10652.
Ртуть йодная, Hgl ТУ 6-09-02-374.
Все реактивы должны быть квалификации х.ч. или ч.д.а.
4.1 . При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами ГОСТ 12.1.007 .
4.2 . Электробезопасность при работе с электроустановками по ГОСТ 12.1.019 .
4.3 . Организация обучения работающих безопасности труда по ГОСТ 12.0.004 .
4.4 . Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009 .
Выполнение измерений может производить химик-аналитик, владеющий техникой фотометрического и спектрофотометрического анализов, изучивший инструкцию по работе с соответствующими приборами.
Измерения проводятся в следующих условиях:
температура окружающего воздуха (20 ± 5) °С;
атмосферное давление (84,0 — 106,7) кПа (630 — 800 мм. рт. ст);
относительная влажность (80 ± 5) %;
напряжение сети (220 ± 10) В;
частота переменного тока (50 ± 1) Гц.
Отбор проб производится в соответствии с требованиями ГОСТ Р 51592-2000 «Вода. Общие требования к отбору проб» G )
Бутыли для отбора и хранения проб воды обезжиривают раствором CMC , промывают водопроводной водой, хромовой смесью, водопроводной водой, а затем 3 — 4 раза дистиллированной водой.
Пробы воды (объем не менее 500 см 3 ) отбирают в стеклянные или полиэтиленовые бутыли, предварительно ополоснув отбираемой водой.
Если определение ионов аммония производят в день отбора пробы, то консервирование не производится. Если проба не будет проанализирована в день отбора, то ее консервируют добавлением 1 см 3 концентрированной серной кислоты на 1 дм 3 . Консервированная проба может храниться не более 2 суток при температуре (3 — 4) ° С. Проба воды не должна подвергаться воздействию прямого солнечного света. Для доставки в лабораторию сосуды с пробами упаковываются в тару, обеспечивающую сохранение и предохраняющую от резких перепадов температуры. При отборе проб составляют сопроводительный документ по форме, в котором указывают:
цель анализа, предполагаемые загрязнители;
должность, фамилия отбирающего пробу, дата.
Подготовку прибора к работе и оптимизацию условий измерения производят в соответствии с рабочей инструкцией по эксплуатации прибора. Прибор должен быть поверен.
Непосредственному применению метода без предварительной отгонки аммиака мешает такое большое количество веществ, что рекомендовать этот метод без отгонки можно для анализа лишь очень немногих вод.
7.4.1 . Определению мешают амины, хлорамины, ацетон, альдегиды, спирты и некоторые другие органические соединения, реагирующие с реактивом Несслера. В их присутствии проводят определение аммиака с отгонкой.
7.4.2 . Определению мешают также компоненты, обуславливающие жесткость воды, железо, сульфиды, хлор, а также мутность.
Мешающее влияние жесткости воды устраняют прибавлением раствора Сегнетовой соли или комплексом (III). Мутные растворы центрифугируют или фильтруют с помощью стеклянной ваты, стеклянного или бумажного фильтра «белая лента», предварительно промытого безаммиачной водой до отсутствия аммиака в фильтре.
Большое количество железа, сульфидов и мутность удаляют с помощью раствора сульфата цинка (см. п. 7.5.8). К 100 см 3 пробы прибавляют 1 см 3 раствора и смесь тщательно перемешивают. Затем рН смеси доводят до 10,5 добавлением 25 %-ного раствора едкого калия или натрия. Проверяют значение рН на рН-метре. После взбалтывания и образования хлопьев осадок отделяют центрифугированием или фильтрованием через стеклянный фильтр (допускается использование бумажного фильтра «белая лента»), предварительно освобожденного от аммиака. Увеличение объема жидкости необходимо учитывать при расчете.
7.4.3 . Мешающее влияние хлора устраняют добавлением раствора тиосульфата или арсенита натрия. Для удаления 0,5 мг хлора достаточно прибавить 1 см 3 одного из указанных растворов (см. п.п. 7.5.11 , 7.5.13 ).
7.4.4 . В присутствии нелетучих органических соединений, например, гуминовых веществ, определение ионов аммония проводят после дистилляции.
7.4.5 . Кальций в концентрациях, превышающих 250 мг/дм 3 , оказывает влияние на установление рН. В этом случае раствор подщелачивают буферным фосфатным раствором и смесь обрабатывают кислотой или щелочью до рН — 7,4 (см. п. 8.2 «Выполнение измерений»).
7.4.6 . Летучие органические соединения, которые мешают определению аммиака в дистилляте, устраняют кипячением слабо подкисленной пробы (см. п. 8.3 «Выполнение измерений»).
7.4.7 . Мутная или цветная вода (при цветности выше 20 ° ) подвергается коагуляции гидроокисью алюминия: к 300 см 3 исследуемой воды прибавляют 2 — 5 см 3 суспензии или 0,5 г сухой окиси алюминия, встряхивают. После 2-часового отстаивания отбирают для анализа прозрачный бесцветный слой.
Если проба воды не осветляется с помощью гидроксида алюминия, ее анализируют после предварительной отгонки (см. п. 8.3 «Выполнение измерений. Определение с перегонкой»).
7.5.1 . Приготовление бидистиллята, не содержащего аммиака
Дважды перегнанную воду пропускают через колонку с катионитом КУ-2 или СБС или: вторично перегоняют дистиллированную воду предварительно подкислив серной кислотой и добавив марганцовокислый калий до четкой малиновой окраски или: упаривают дистиллированную воду до 1/4 объема, после добавления двууглекислого натрия (0,1 — 0,5 г на 1 дм 3 ). Полученную воду проверяют на наличие аммиака реактивом Несслера и используют для приготовления реактивов и разбавления проб.
7.5.2 . Приготовление основного раствора хлористого аммония
2 ,9650 г аммония хлористого, подготовленного по ГОСТ 4212 , помещают в стакан, растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу на 1000 см 3 , а затем доводят до метки.
1 см 3 раствора содержит 1 мг NH 4 + . Приготовленный раствор хранят в банке из темного стекла в течение года.
7.5.3 . Приготовление рабочего раствора хлористого аммония
Раствор готовят в день проведения анализа, разбавлением основного стандартного раствора безаммиачной водой.
1 см 3 раствора содержит 0,005 мг NН4 + .
При наличии ГСО: раствор готовят в соответствии с прилагаемой к образцу инструкцией.
1 см 3 раствора должен содержать 0,005 мг N Н4 + .
7.5.4 . Приготовление боратного буферного раствора, рН = 9,5
К 500 см раствора 0,025 М тетрабората натрия приливают 88 см 3 0,1 М раствора гидроокиси натрия и разбавляют до 1 дм 3 безаммиачной водой. Хранят в течение 3 мес.
7.5.5 . Приготовление тетрабората натрия, 0,025 М водный раствор
9 ,5 г тетрабората натрия ( Na 2 B 4 О ∙ 10Н2О) помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в колбу на 1000 см 3 , а затем доводят до метки. Хранят в течение 3 мес.
7.5.6 . Приготовление фосфатного буферного раствора рН = 7,4
14 ,3 г безводного однозамещенного фосфорнокислого калия и 68,8 г безводного двузамещенного фосфорнокислого калия помещают в стакан, растворяют в небольшом количестве безаммиачной воды, не содержащей аммиака и аммонийных солей, переносят в мерную колбу на 1 дм 3 , и доводят до метки этой же водой.
Используют выпускаемый реактив по ТУ 6-09-2089.
В случае отсутствия готовят в лабораторных условиях из окиси ртути (II) одним из указанных методов.
Исходный материал: ртуть хлорная, ртуть йодная.
HgCl 2 (ртуть хлорная): готовят растворением окиси ртути в разбавленной соляной кислоте: HgO + 2HCl = HgCl 2 + Н2O
HgI 2 (ртуть йодная): получают при взаимодействии хлорной ртути с йодистым калием: HgCl 2 + 2KI = HgI 2 + 2KCl (Карякин Ю.В., Ангелов И.И. «Чистые химические вещества». М., Химия, 1974, с. 309 — 310, 314).
50 г иодида калия помещают в стакан, растворяют в 50 см 3 безаммиачной воды. Отдельно 30 г хлорида ртути ( II ) помещают в стакан, растворяют в 150 см 3 нагретой до кипения безаммиачной воды. Горячий раствор хлорида ртути приливают к раствору иодида калия до появления не исчезающего при перемешивании красного осадка. Затем фильтруют через стеклянный фильтр или слой прокаленного асбеста и к фильтрату прибавляют раствор 150 г едкого кали в 300 см 3 безаммиачной воды.
Разбавив полученный раствор до 1 дм 3 , вводят в него еще 5 см 3 насыщенного раствора хлорида ртути (II ) и оставляют в темном месте до полного осветления. Хранят в темном месте, в склянке, закрытой корковой пробкой. При употреблении отбирают пипеткой прозрачную жидкость не взмучивая осадка со дна склянки.
Или: 100 г безводного иодида ртути ( II ) и 70 г безводного иодида калия помещают в стакан, растворяют в небольшом количестве безаммиачной воды, полученную смесь медленно, при непрерывном перемешивании переносят в охлажденный раствор, полученный при растворении 160 г едкого натра в 500 см 3 безаммиачной воды. Полученную смесь разбавляют безаммиачной водой до 1 дм 3 .
7.5.8 . Приготовление водного раствора сульфата цинка
100 г сульфата цинка помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой.
10 г гидроксида натрия помещают в стакан, растворяют в 60 см 3 безаммиачной воды. К полученному раствору добавляют 50 г трилона Б, переносят в мерную колбу на 100 см 3 и доводят до метки безаммиачной водой.
7.5.10 . Приготовление раствора калия натрия виннокислого (сегнетова соль)
50 г KNaC 4 H 4 O 6 ∙ 4Н2О помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 100 см 3 , доводят до метки бидистиллированной водой, прибавляют 0,2 — 0,5 см 3 реактива Несслера. Раствор можно применять после осветления.
7.5.11 . Приготовление водного раствора арсенита натрия
1 г мышьяковистого натрия помещают в стакан, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой.
7.5.12 . Приготовление водного раствора сульфата натрия
0 ,9 г сернистокислого натрия помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой.
7.5.13 . Приготовление водного раствора тиосульфата натрия
3 ,5 г серноватистокислого натрия помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой.
40 г борной кислоты помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой.
7.5.15 . Приготовление гидроокиси алюминия, суспензии для коагуляции
125 г алюмокалиевых квасцов AIK (SО4)2 ∙ 12Н2О помещают в стакан, растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу на 1 дм 3 , доводят до метки дистиллированной водой, нагревают до 60 °С и постепенно прибавляют 55 см 3 концентрированного раствора аммиака при постоянном перемешивании. Дают смеси постоять около 1 часа, промывают осадок гидроксида алюминия многократной декантацией дистиллированной водой до удаления хлоридов, нитритов, нитратов и аммиака.
7.5.16 . Приготовление 1 М водного раствора серной кислоты
27 ,3 см 3 серной кислоты пл. 1,84 г/см 3 вносят небольшими порциями при перемешивании в 150 — 200 см 3 дистиллированной воды, переносят в мерную колбу вместимостью 1 дм 3 и доводят до метки дистиллированной водой.
7.5.17 . Приготовление 40 %-ного раствора гидроокиси натрия
40 г гидроокиси натрия помещают в стакан, растворяют в 60 см 3 безаммиачной воды.
7.5.18 . Приготовление 15 %-ного раствора гидроокиси натрия
15 г гидроокиси натрия помещают в стакан, растворяют в 85 см 3 безаммиачной воды.
7.5.19 . Приготовление 1 М раствора гидроокиси натрия
40 г гидроокиси натрия помещают в стакан, растворяют в небольшом количестве безаммиачной воды, переносят в мерную колбу на 1 дм 3 и доводят до метки безаммиачной водой. Хранят под защитой от контакта с воздухом.
К 10 см 3 пробы прибавляют несколько кристалликов сегнетовой соли и 0,5 см 3 реактива Несслера. Желтое окрашивание раствора, помутнение или выпадение желто-коричневого осадка указывает на присутствие ионов аммония. При повышенном содержании органических веществ, особенно гуминовых кислот, вызывающих усиление коричневой окраски после подщелачивания, проводят параллельный опыт, добавив к пробе сегнетову соль, а вместо реактива Несслера — 0,5 см 3 15 %-ного раствора гидроксида натрия.
К 50 см 3 первоначальной или осветленной пробы, или к меньшему ее объему, доведенному до 50 см 3 безаммиачной водой, прибавляют 1 — 2 капли раствора сегнетовой соли или комплексона III и смесь тщательно перемешивают. При анализе очень жестких вод количество добавляемого раствора сегнетовой соли или комплексона III увеличивается до 0,5 — 1,0 см 3 . Затем добавляют 1 см 3 реактива Несслера и снова перемешивают. Через 10 минут измеряют оптическую плотность. Окраска смеси устойчива в течение 30 мин. Из величины оптической плотности вычитают оптическую плотность холостого опыта. Если необходимо, вычитают и оптическую плотность пробы, к которой вместо реактива Несслера добавляют 1 см 3 15 %-ного раствора едкого натра и по графику находят содержание ионов аммония.
При анализе окрашенных проб, а также в присутствии мешающих органических соединений производят предварительную отгонку аммиака из исследуемой воды.
Отгонку аммиака из проб природных и сточных вод, содержащих легко гидролизуемые органические соединения, проводят при рН ÷ 7,4 добавляя к пробе фосфатный буферный раствор; в присутствии цианидов и большинства азотсодержащих органических соединений следует использовать боратный буферный раствор (рН ÷ 9,5). При анализе сточных вод, содержащих большие количества фенолов (воды коксохимических, газогенераторных предприятий) к пробе воды добавляют 40 %-ный раствор гидроксида натрия. Если наряду с фенолами присутствуют вещества, гидролизующиеся в щелочной среде, то отгонку надо провести дважды: сначала при рН ÷ 7,4 собирая отгон в разбавленный раствор серной кислоты, затем подщелочить этот отгон до сильнощелочной реакции.
Для поглощения аммиака применяют растворы борной или серной кислот или безаммиачную воду.
Перегонку исследуемых проб проводят в комнате, воздух которой не содержит аммиака.
В колбу для отгона помещают 400 см 3 анализируемой пробы воды (или меньший объем, доведенный до 400 см 3 безаммиачной водой). Если проба воды содержит большое количество взвешенных веществ или нефтепродуктов, ее предварительно фильтруют через фильтр «белая лента». При необходимости пробу воды дехлорируют одним из реагентов, рекомендованных в п. 7.4.3. Если надо, нейтрализуют пробу (до рН ÷ 7) 1 М раствором серной кислоты или гидроксида натрия. Затем приливают 25 см 3 буферного раствора (рН ÷ 7,4 или 9,5 в зависимости от предполагаемых загрязнений) или 20 см 3 40 %-ного раствора гидроокиси натрия при анализе фенольных вод. В приемник наливают 50 см 3 поглощающего раствора и устанавливают объем жидкости так, чтобы конец холодильника был погружен в нее, добавляя при необходимости безаммиачную воду. Отгоняют примерно 300 см 3 жидкости, отгон количественно переносят в мерную колбу на 500 см 3 , измеряют рН полученного отгона (по рН-метру) и при необходимости доводят рН раствора до 6,0, затем разбавляют до метки безаммиачной водой.
В 50 см 3 аликвотной части определяют содержание ионов аммония, как указано в п. 8.2. При измерении оптической плотности используют кюветы толщиной слоя 1 — 5 см в зависимости от содержания ионов аммония в растворе.
В мерные колбы вместимостью 50 см 3 вносят 0,0; 0,5; 1,0; 2,0; 4,0; 6,0; 8,0; 10,0; . 40,0 см 3 рабочего стандартного раствора аммония (п. 7.5.3), доводят до метки безаммиачной водой. Полученную шкалу растворов с содержанием 0,0; 0,0025; 0,005; 0,01; 0,02; 0,03; 0,04; 0,05; . 0,2 мг NH 4 + обрабатывают описанным выше (п. 8.2) способом.
График строят методом наименьших квадратов в координатах оптическая плотность — содержание ионов аммония (мг); вводят поправку на холостой опыт.
Для растворов с содержанием 0,0 — 0,03 мг ионов аммония строят график, используя кюветы толщиной слоя 5 см; для растворов, содержащих 0,03 — 0,20 мг NH 4 + — график с использованием кюветы с толщиной слоя 1 см.
Контроль стабильности градуировочной характеристики проводят не реже одного раза в квартал или при смене партий реактивов. Средствами контроля являются вновь приготовленные образцы для градуировки (не менее 3 образцов из приведенных в п. 8.4).
Градуировочную характеристику считают стабильной при выполнении для каждого образца для градуировки следующего условия:
где X — результат контрольного измерений массовой концентрации ионов аммония в образце для градуировки;
С — аттестованное значение массовой концентрации ионов аммония в образце для градуировки;
s Rл _ среднеквадратическое отклонение внутрилабораторной прецизионности, установленное при реализации методики в лаборатории.
Примечание. Допустимо среднеквадратическое отклонение внутрилабораторной прецизионности при внедрении методики в лаборатории устанавливать на основе выражения: s Rл = 0,84 s r , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Значения s r приведены в таблице 1.
Если условие стабильности градуировочной характеристики не выполняется только для одного образца для градуировки, необходимо выполнить повторное измерение этого образца с целью исключения результата содержащего грубую погрешность.
Если градуировочная характеристика нестабильна, выясняют причины и повторяют контроль с использованием других образцов для градуировки, предусмотренных методикой. При повторном обнаружении нестабильности градуировочной характеристики строят новый градуировочный график.
Содержание ионов аммония NH 4 + в мг/дм 3 вычисляют по формуле:
где С — содержание ионов аммония, найденное по калибровочному графику, мг,
V — объем пробы, взятой для анализа, см 3 ;
n = 1 при прямом определении ионов аммония;
n = 10 при определении с предварительной отгонкой аммиака (т.к. для анализа используется 1/10 отгона).
За результат анализа Хср принимают среднее арифметическое значение двух параллельных определений Х1 и Х2.
для которых выполняется следующее условие:
где r — предел повторяемости, значения которого приведены в таблице 2.
Значения предела повторяемости при вероятности Р = 0,95
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных определений), r, %
При невыполнении условия (1) могут быть использованы методы проверки приемлемости результатов параллельных определений и установления окончательного результата согласно раздела 5 ГОСТ Р ИСО 5725-6.
Расхождение между результатами анализа, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата анализа, и в качестве окончательного может быть использовано их среднее арифметическое значение. Значения предела воспроизводимости приведены в таблице 3.
Значения предела воспроизводимости при вероятности Р = 0,95
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R, %
При превышении предела воспроизводимости могут быть использованы методы оценки приемлемости результатов анализа согласно раздела 5 ГОСТ Р ИСО 5725-6.
10.1 . Результат анализа Хср в документах, предусматривающих его использование, может быть представлен в виде: Хср ± D , Р = 0,95
где D — показатель точности методики.
Значение D рассчитывают по формуле: D = 0,01 ∙ δ ∙ X ср . Значение δ приведено в таблице 1 .
Допустимо результат анализа в документах, выдаваемых лабораторией, представлять в виде: Хср ± D л , Р = 0,95, при условии D л D ,
где Хср— результат анализа, полученный в соответствии с прописью методики;
± D л — значение характеристики погрешности результатов анализа, установленное при реализации методики в лаборатории, и обеспечиваемое контролем стабильности результатов анализа.
Примечание. При представлении результата анализа в документах, выдаваемых лабораторией, указывают:
— количество результатов параллельных определений, использованных для расчета результата анализа;
— способ определения результата анализа (среднее арифметическое значение или медиана результатов параллельных определений).
10.2 . В том случае, если массовая концентрация ионов аммония в анализируемой пробе превышает верхнюю границу диапазона, то допускается разбавление пробы таким образом, чтобы массовая концентрация ионов аммония соответствовала регламентированному диапазону.
Результат анализа Хср в документах, предусматривающих его использование, может быть представлен в виде: Хср± D ‘, Р = 0,95 , где ± D ‘ — значение характеристики погрешности результатов анализа, откорректированное на величину погрешности взятия аликвоты.
Контроль качества результатов анализа при реализации методики в лаборатории предусматривает:
— оперативный контроль процедуры анализа (на основе оценки погрешности при реализации отдельно взятой контрольной процедуры);
— контроль стабильности результатов анализа (на основе контроля стабильности среднеквадратического отклонения повторяемости, среднеквадратического отклонения внутрилабораторной прецизионности, погрешности).
Оперативный контроль процедуры анализа проводят путем сравнения результата отдельно взятой контрольной процедуры Кк с нормативом контроля К.
Результат контрольной процедуры Кк рассчитывают по формуле:
где х ‘ ср — результат анализа массовой концентрации ионов аммония в пробе с известной добавкой — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 9.
X ср — результат анализа массовой концентрации ионов аммония в исходной пробе — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 9 .
Норматив контроля К рассчитывают по формуле
где — значения характеристики погрешности результатов анализа, установленные в лаборатории при реализации методики, соответствующие массовой концентрации ионов аммония в пробе с известной добавкой и в исходной пробе соответственно.
Примечание. Допустимо характеристику погрешности результатов анализа при внедрении методики в лаборатории устанавливать на основе выражения: D л = 0,84 D , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Процедуру анализа признают удовлетворительной, при выполнении условия:
При невыполнении условия (2) контрольную процедуру повторяют. При повторном невыполнении условия (2) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры по их устранению.
Оперативный контроль процедуры анализа проводят путем сравнения результата отдельно взятой контрольной процедуры Кк с нормативом контроля К.
Результат контрольной процедуры Кк рассчитывают по формуле:
где Сср — результат анализа массовой концентрации ионов аммония в образце для контроля — среднее арифметическое двух результатов параллельных определений, расхождение между которыми удовлетворяет условию (1) раздела 9;
С — аттестованное значение образца для контроля.
Норматив контроля К рассчитывают по формуле:
где ± D л — характеристика погрешности результатов анализа, соответствующая аттестованному значению образца для контроля.
Примечание. Допустимо характеристику погрешности результатов анализа при внедрении методики в лаборатории устанавливать на основе выражения: D л = 0,84 D , с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов анализа.
Процедуру анализа признают удовлетворительной, при выполнении условия:
При невыполнении условия (3) контрольную процедуру повторяют. При повторном невыполнении условия (3) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры по их устранению.
Периодичность оперативного контроля процедуры анализа, а также реализуемые процедуры контроля стабильности результатов анализа регламентируют в Руководстве по качеству лаборатории.
источник
Настоящий нормативный документ устанавливает методику количественного химического анализа различных типов вод с целью определения суммарного содержания аммиака и аммоний-ионов (далее — аммония) фотометрическим методом с реактивом Несслера. Методика распространяется на следующие объекты анализа: воды питьевые, в том числе расфасованные в емкости; воды природные пресные подземных источников водоснабжения; воды сточные производственные, хозяйственно-бытовые, ливневые и очищенные. Методика может быть использована для анализа талых, технических вод и проб снежного покрова.
Методику не рекомендуется использовать для анализа проб воды поверхностных водоемов, т.к. природные гуминовые вещества мешают определению аммония. Процедура осаждения гуминовых веществ в щелочной среде не позволяет устранить мешающее влияние полностью, что приводит к завышению результатов определения аммония в диапазоне массовых концентраций от 0,1 до 1 мг/дм 3 . Для анализа природных вод поверхностных водоемов рекомендуется использовать другой метод анализа.
Диапазон измерений массовых концентраций аммония составляет от 0,1 до 100 мг/дм 3 .
Примечание — Если массовая концентрация аммония в пробе превышает верхнюю границу указанного диапазона , то допускается разбавление пробы таким образом, чтобы массовая концентрация аммония в разбавленной пробе соответствовала диапазону.
При массовой концентрации аммония в анализируемой пробе свыше 3,0 мг/дм 3 анализ выполняют с разбавлением пробы.
Мешающее влияние мутности и цветности проб устраняют путем проведения процедуры осаждения раствором сульфата меди в щелочной среде.
Мешающее влияние фенолов, сероводорода и сульфидов и некоторых органических веществ, например, аминов, ацетона, альдегидов и спиртов, устраняется путем отгонки аммиака из щелочного раствора.
Мешающее влияние солей кальция и магния устраняют добавлением раствора калия-натрия виннокислого.
При взаимодействии активного остаточного хлора с аммоний-ионами образуются хлорамины, которые мешают определению аммония. Мешающее влияние активного хлора устраняют добавлением эквивалентного количества раствора серноватистокислого натрия.
При невозможности устранения мешающих влияний с помощью процедур пробоподготовки, предусмотренных настоящим нормативным документом, рекомендуется выполнять анализ с использованием другого метода анализа.
Блок-схема проведения анализа приведена в приложении 1.
ГОСТ 12.0.004-90 Система стандартов безопасности труда. Организация обучения безопасности труда. Общие положения
ГОСТ 12.1.004-91 Система стандартов безопасности труда. Пожарная безопасность. Общие требования
ГОСТ 12.1.007-76 Система стандартов безопасности труда. Вредные вещества. Классификация и общие требования безопасности
ГОСТ 12.4.009-83 Система стандартов безопасности труда. Пожарная техника для защиты объектов. Основные виды. Размещение и обслуживание
ГОСТ 17.1.5.05-85 Охрана природы. Гидросфера. Общие требования к отбору проб поверхностных и морских вод, льда и атмосферных осадков
ГОСТ 1770-74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Общие технические условия
ГОСТ 4165-78 Реактивы. Медь (II) сернокислая 5-водная. Технические условия
ГОСТ 4232-74 Реактивы. Калий йодистый. Технические условия
ГОСТ 4328-77 Реактивы. Натрия гидроокись. Технические условия
ГОСТ 5845-79 Реактивы. Калий-натрий виннокислый 4-водный. Технические условия
ГОСТ 6709-72 Вода дистиллированная. Технические условия
ГОСТ 9656-75 Реактивы. Кислота борная. Технические условия
ГОСТ 14262-78 Кислота серная особой чистоты. Технические условия
ГОСТ 14919-83 Электроплиты, электроплитки и жарочные электрошкафы бытовые. Общие технические условия
ГОСТ 18190-72 Вода питьевая. Методы определения содержания остаточного активного хлора
ГОСТ 24363-80 Реактивы. Калия гидроокись. Технические условия
ГОСТ 25336-82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 27384-2002 Вода. Нормы погрешностей измерений показателей состава и свойств
ГОСТ 28311-89 Дозаторы медицинские лабораторные. Общие технические требования и методы испытаний
ГОСТ 29169-91 Посуда лабораторная стеклянная. Пипетки с одной отметкой
ГОСТ 29227-91 Посуда лабораторная стеклянная. Пипетки градуированные. Часть 1. Общие требования
ГОСТ Р 12.1.019-2009 Система стандартов безопасности труда. Электробезопасность. Общие требования и номенклатура видов защиты
ГОСТ Р ИСО 5725-6-2002 Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности
ГОСТ Р 51592-2000 Вода. Общие требования к отбору проб
ГОСТ Р 52501-2005 Вода для лабораторного анализа. Технические условия
ГОСТ Р 53228-2008 Весы неавтоматического действия. Часть 1. Метрологические и технические требования. Испытания
Примечание — Если ссылочный стандарт заменен (изменен), то следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
Настоящая методика обеспечивает получение результатов анализа с погрешностями, не превышающими значений, приведенных в таблице 1. Приписанные погрешности измерений не превышают нормы погрешностей, установленные ГОСТ 27384.
Таблица 1 — Диапазон измерения, значение показателей повторяемости, воспроизводимости и точности
Показатель повторяемости (стандартное отклонение повторяемости), s r, %
Показатель воспроизводимости (стандартное отклонение воспроизводимости) s R, %
Показатель точности (границы относительной погрешности при Р = 0,95), ± δ, %
источник
Требования, предъявляемые к качеству воды, могут быть самыми различными и определяются её целевым назначением. Для оценки качества пластовых, природных и сточных вод их образцы подвергают анализу. На основании результатов анализа делаются выводы о пригодности воды для конкретного вида потребления, возможности применения тех или иных методов очистки. Анализы подземных вод позволяют прогнозировать сопутствующие месторождения полезных ископаемых. При анализе вод для характеристики их свойств определяют химические, физические и бактериологические показатели. Основными показателями, определяющими пригодность воды для определенной отрасли народного хозяйства, являются химические, так как физические (содержание взвешенных частиц, температура, цвет, запах, плотность, сжимаемость, вязкость, поверхностное натяжение) и бактериологические (наличие бактерий) показатели зависят от химического состава воды.
К химическим показателям качества воды относятся:
состав растворенных газов.
Общее солесодержание характеризует присутствие в воде минеральных и органических примесей, количество этих примесей в виде общей минерализации, сухого и плотного остатков. Общая минерализация представляет собой сумму всех найденных в воде анализом катионов и анионов. Минерализацию выражают в миллиграмм-эквивалентах солей, находящихся в I л воды, или в процентах, то есть числом граммов растворенных веществ, содержащихся в 100 г раствора. Сухим остатком называется суммарное количество нелетучих веществ, присутствующих в воде во взвешенном, коллоидном и растворенном состоянии, выраженное в мг/л. Сухой остаток определяют путем выпаривания пробы воды, последующего высушивания при 105 о С и взвешивания. Плотный остаток – это сухой остаток, определенный из профильтрованной пробы воды. Следовательно, разница двух показателей соответствует содержанию взвешенных веществ пробы. Если сухой остаток прокалить при температуре 500-600 о С, то масса его уменьшится и получится остаток, называемый золой. Уменьшение массы происходит за счет сгорания органических веществ, удаления кристаллизационной воды, разложения карбонатов. Потери при прокаливании приближенно относят за счет органических примесей.
Жесткость воды обусловливается наличием в ней ионов Са 2+ и Mg 2+ . Для большинства производств жесткость воды является основным показателем её качества. В жесткой воде плохо пенится мыло. При нагревании и испарении жесткой воды образуется накипь на стенках паровых котлов, труб, теплообменных аппаратов, что ведет к перерасходу топлива, коррозии металлов и авариям.
Жесткость количественно выражается числом миллиграмм-эквивалентов ионов кальция и магния в 1 л воды (мг-экв/л); 1 мг-экв/л жесткости соответствует содержанию в воде 20,04 мг/л ионов Са 2+ или
12,16 мг/л ионов Mg 2 + . Различают жесткость общую, карбонатную и некарбонатную.
Карбонатная жесткость связана с присутствием в воде в основном гидрокарбонатов и карбонатов кальция и магния, которые при кипячении воды переходят в нерастворимые средние или основные соли и выпадают в виде плотного осадка:
Таким образом, при кипячении карбонатная жесткость устраняется. Поэтому она называется также временной жесткостью. Следует сказать, что при переходе HCO3 – в CO32 – и при выпадении карбонатов кальция и магния в воде остается некоторое количество ионов Са 2+ , Mg 2+ , CO32 – , соответствующее произведению растворимости СаСО3 и (MgOH)2CO3. В присутствии посторонних ионов растворимость этих соединений повышается.
Некарбонатная (постоянная) жесткость не разрушается кипячением. Она обусловливается присутствием в воде кальциевых и магниевых солей сильных кислот, главным образом сульфатов и хлоридов.
Общаяжесткость воды представляет собой сумму карбонатной и некарбонатной жесткости и обусловливается суммарным содержанием в воде растворенных солей кальция и магния. По величине общей жесткости принята следующая классификация природных вод:
Если известны концентрации (мг/л) в воде Ca 2+ , Mg 2+ и HCO3 – , то жесткость рассчитывается по следующим формулам:
Общая жесткость 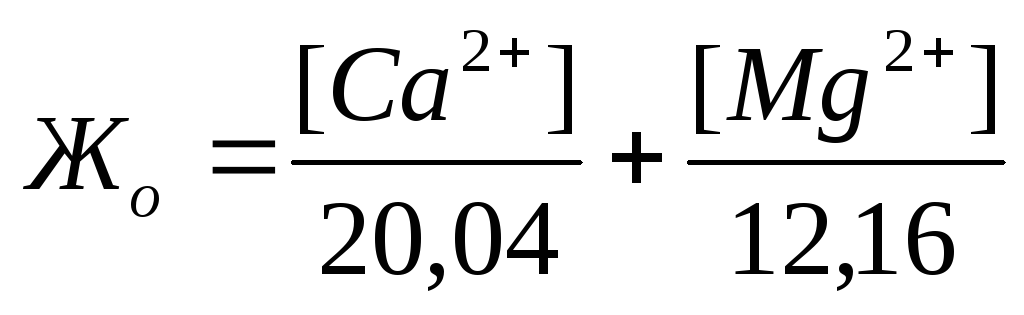
Карбонатная жесткость равна концентрации (мг/л) [HCO3– ]; в случае, если содержание ионов кальция и магния в воде выше, чем количество гидрокарбонатов:
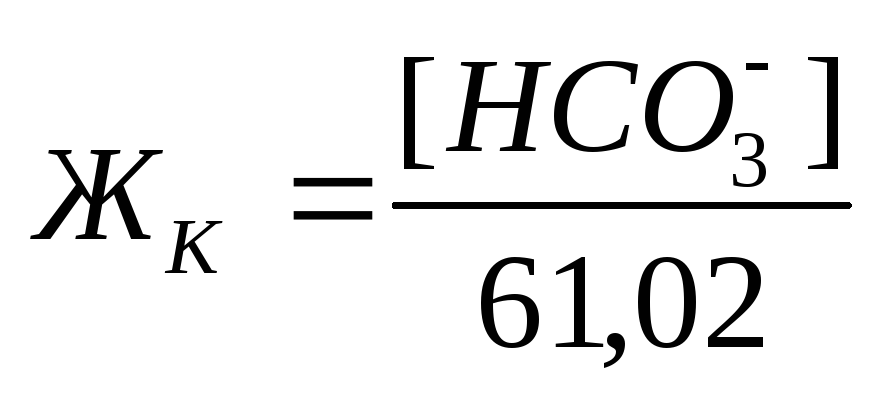 , где 61,02 – эквивалентная масса иона HCO3 – .
, где 61,02 – эквивалентная масса иона HCO3 – .
Если же количество гидрокарбонатов в воде превышает содержание ионов кальция и магния, то карбонатная жесткость соответствует общей жесткости. Разность между общей и карбонатной жесткостью составляет некарбонатную жесткость: ЖНК= ЖО– ЖК . Следовательно, ЖНК – это содержание Ca 2+ и Mg 2 + , эквивалентное концентрации всех остальных анионов, в том числе и некомпенсированных гидрокарбонатов.
Окисляемость характеризует содержание в воде восстановителей, к которым относятся органические и некоторые неорганические (сероводород, сульфиты, соединения двухвалентного железа и др.) вещества. Величина окисляемости определяется количеством затраченного окислителя и выражается числом миллиграммов кислорода, необходимого для окисления веществ, содержащихся в 1 л воды. Различают общую и частичную окисляемость. Общую окисляемость определяют обработкой воды сильным окислителем – бихроматом калия K2Cr2O7 или йодатом калия KIO3. Частичную окисляемость определяют по реакции с менее сильным окислителем – перманганатом калия КMnO4. По этой реакции окисляются только сравнительно легко окисляющиеся вещества.
Для полного окисления содержащихся в воде органических веществ, при котором происходят превращения по схеме
требуется количество кислорода (или окислителя в расчете на кислород), называемое химическим потреблением кислорода (ХПК) и выражаемое в мг/л.
При любом методе определения ХПК вместе с органическими веществами окисляются и неорганические восстановители, содержащиеся в пробе. Тогда содержание неорганических восстановителей в пробе определяют отдельно специальными методами и результаты этих определений вычитают из найденного значения ХПК.
Реакция среды характеризует степень кислотности или щелочности воды. Концентрация водородных ионов природных вод зависит главным образом от гидролиза солей, растворенных в воде, количества растворенных угольной кислоты и сероводорода, содержания различных органических кислот. Обычно для большинства природных вод величина рН изменяется в пределах 5,5-8,5. Постоянство рН природных вод обеспечивается наличием в ней буферных смесей. Изменение значения рН свидетельствует о загрязнении природной воды сточными водами.
Определение иона Cl – . В основу определения иона хлора положен аргентометрический метод Мора. Принцип анализа заключается в том, что при прибавлении к воде раствора AgNO3 образуется белый осадок хлорида серебра:
Определение хлорид-ионов ведут в интервале рН = 6,5 ÷ 10, чтобы одновременно с AgCl не выпадал осадок Ag2CO3. Проведению определения Сl – мешает наличие в воде ионов брома, йода, сероводорода, от которых освобождаются предварительной обработкой воды.
Определение иона SO42– . Метод определения сульфат-ионов основан на малой растворимости сульфата бария, количественно выпадающего в кислой среде при добавлении к воде раствора хлорида бария: Ba 2+ + SO42– = BaSO4↓
По массе образовавшегося осадка рассчитывают содержание иона SO42– .
Определение ионов CO32– и HCO3– . Эти ионы определяют титрованием пробы воды растворами серной или соляной кислот последовательно с индикаторами фенолфталеином и метилоранжем. Реакция нейтрализации протекает в две стадии.
Первые порции кислоты вступают в реакции с карбонат-ионом, образуя гидрокарбонат-ион:
Окраска фенолфталеина при рН = 8,4 переходит из розовой в бесцветную, что совпадает с таким состоянием раствора, когда в нем остаются лишь гидрокарбонаты. По количеству кислоты, пошедшей на титрование, рассчитывают содержание карбонат-иона. Расход кислот на титрование с фенолфталеином эквивалентен содержанию половины карбонатов, т.к. последние нейтрализуются только наполовину до HCO3 – . Поэтому общее количество CO32 – эквивалентно удвоенному количеству кислоты, затраченной на титрование. При дальнейшем титровании в присутствии метилоранжа происходит реакция нейтрализации гидрокарбонатов:
Метилоранж меняет окраску при pH = 4,3, т.е. в момент, когда в растворе остается только свободный диоксид углерода.
При расчете содержания ионов HCO3 – в воде следует из количества кислоты, пошедшей на титрование с метилоранжем, вычесть количество кислоты, идущей на титрование с фенолфталеином. Общее количество кислоты, затраченной на нейтрализацию ионов ОН – , СО32– и НСО3– , характеризует общую щелочность воды. Если рН воды ниже 4,3, то её щелочность равна нулю.
Определение ионов Ca 2+ , Mg 2+ . Имеется несколько методов обнаружения и определения содержания ионов Са 2+ и Mg 2+ . При добавлении в воду оксалата аммония (NH4)2C2O4 в случае присутствия ионов кальция образуется белый осадок оксалата кальция:
После отделения осадка оксалата кальция в воде можно определить ионы Mg 2+ с помощью раствора гидрофосфата натрия Na2HPO4 и аммиака. При наличии иона Mg 2 + образуется мелкокристаллический осадок соли магния:
Полученные осадки прокаливают и взвешивают. На основании полученных результатов вычисляется величина кальциевой и магниевой жесткости.
Наиболее быстрым и точным методом определения Са 2 + и Mg 2 + является комплексонометрический метод, основанный на способности двунатриевой соли этилендиаминотетрауксусной кислоты (трилон Б)
N
 aOOCCH2 CH2COONa
aOOCCH2 CH2COONa
 N––CH2––CH2––N
N––CH2––CH2––N
образовывать с ионами кальция и магния прочные комплексные соединения.
При титровании пробы воды трилоном Б происходит последовательное связывание в комплекс сначала ионов кальция, а затем ионов магния. Содержание ионов кальция определяют, титруя воду в присутствии индикатора — мурексида. Мурексид образует с ионами кальция малодиссоциированное комплексное соединение, окрашенное в малиновый цвет.
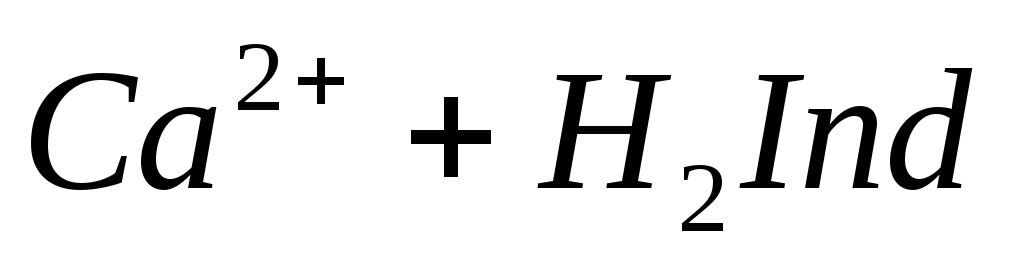

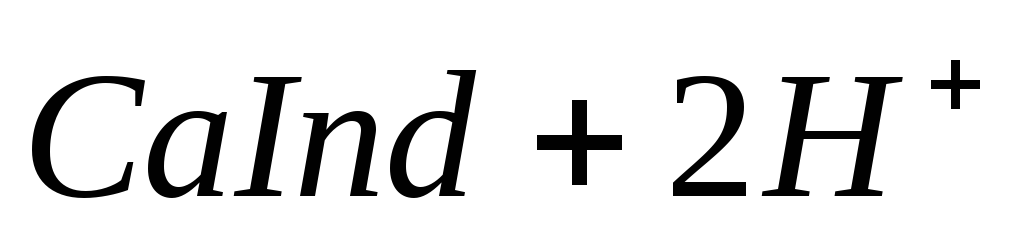
Ионы магния не дают комплекса с мурексидом. Трилон Б извлекает Са 2+ из его растворимого комплекса с мурексидом, вследствие чего окраска раствора, изменяется на сиреневую:
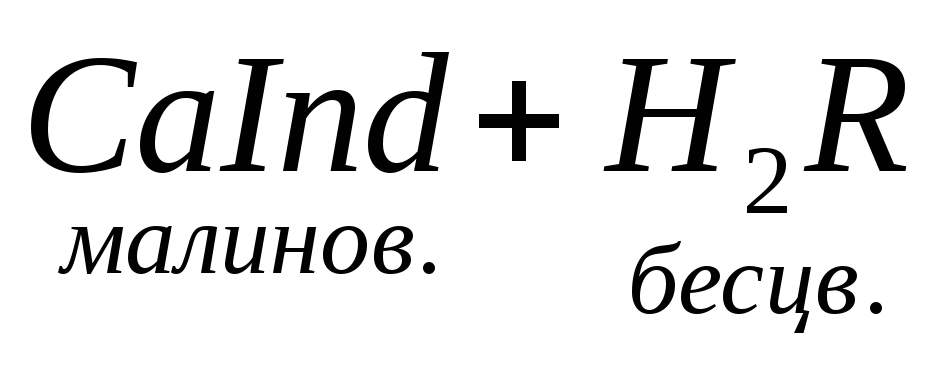

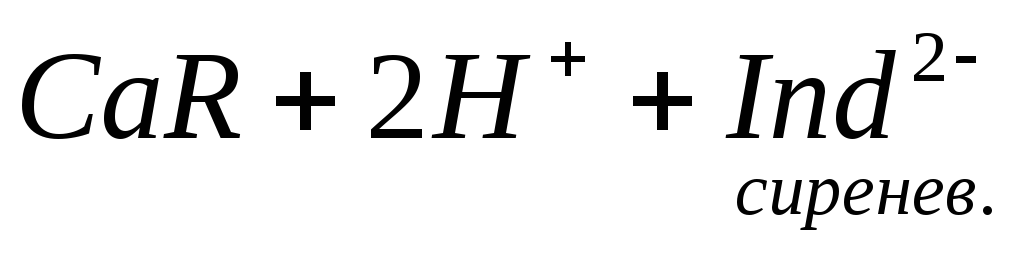
По количеству трилона Б, расходуемого на титрование, определяют содержание Са 2 + . Титрованием пробы воды трилоном Б в присутствии индикатора хромогена черного определяют суммарное содержание Са 2 + и Mg 2 + , то есть общую жесткость воды. Вода, содержащая Са 2 + и Mg 2 + , в присутствии хромогена черного окрашивается в красный цвет вследствие образования комплекса с Mg 2 + . При титровании воды в точке эквивалентности происходит изменение цвета на синий вследствие протекания следующей реакции:
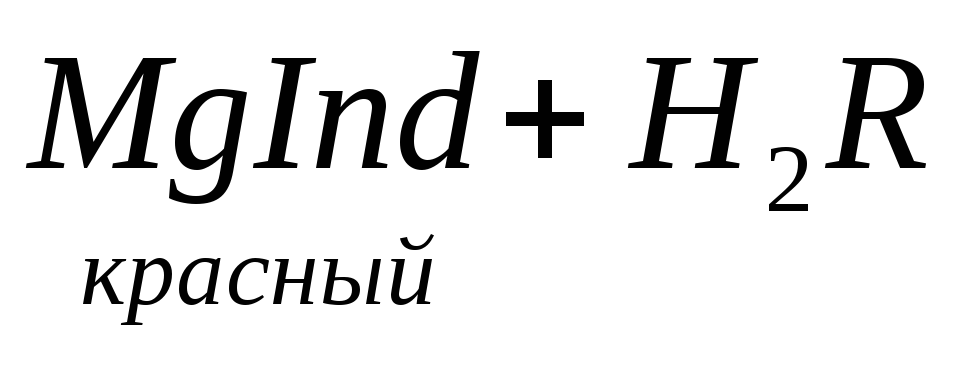

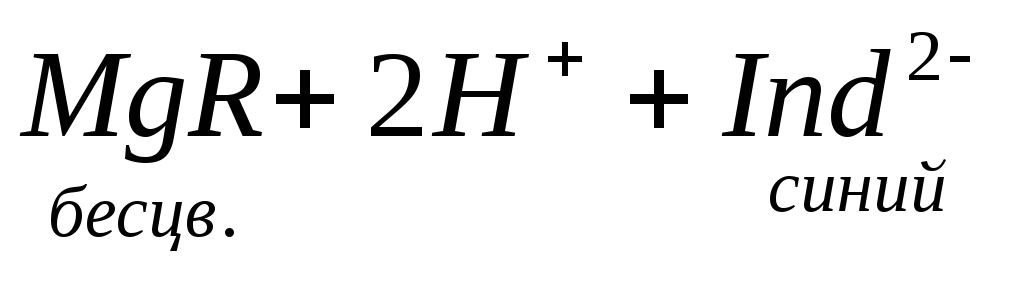
Содержание Mg 2+ вычисляют по разности между общим содержанием (Са 2+ + Mg 2+ ) и содержанием Са 2 + . Трилонометрическое определение каждого иона производится при том значении рН, при котором этот ион образует с трилоном Б соединение более прочное, чем с индикатором. Для поддержания заданного значения рН к титруемому раствору добавляют буферные растворы. Кроме того, поддержание заданной величины рН обеспечивает определенную окраску индикатора. Общую жесткость воды определяют при рН > 9, кальциевую – при рН = 12.
Определение ионов Na + , K + . Производится вычислением по разности между суммой мг-экв найденных анионов и катионов, поскольку вода электронейтральна:
С достаточно высокой точностью все присутствующие в воде катионы можно определить эмиссионной спектроскопией сухого остатка.
Растворенные в воде газы определяют химическими методами или газовой хроматографией.
Определение диоксида углерода производят титрованием пробы воды щелочью в присутствии индикатора–фенолфталеина:
Определение растворенного кислорода производится йодометрическим методом.
Для анализа в пробу воды поcледовательно добавляют раствор хлорида марганца и щелочной раствор йодида калия. Метод основан на окислении свежеполученного гидроксида двухвалентного марганца содержащимся в воде кислородом:
Количество образовавшегося в воде бурого осадка гидроксида четырехвалентного марганца эквивалентно количеству растворенного кислорода. При последующем добавлении к пробе соляной или серной кислоты четырехвалентный марганец вновь восстанавливается до двухвалентного, окисляя при этом йодид калия. Это приводит к выделению свободного йода, эквивалентного содержанию четырехвалентного марганца, или, что то же самое, растворенного кислорода в пробе:
Выделившийся свободный йод определяется количественно путем титрования раствором тиосульфата натрия:
I2+ 2Na2S2O3 2NaI + Na2S4O6
2NaI + Na2S4O6
Йодометрический метод определения растворенного кислорода неприменим для вод, содержащих сероводород, так как сероводород вступает во взаимодействие с йодом и занижает результат. Во избежание этой ошибки предварительно связывают содержащийся в пробе сероводород в соединение, не препятствующее нормальному течению реакции. Для этой цели обычно используют хлорид ртути (II):
Определение H2S. Прежде чем приступить к количественному определению сероводорода, определяют его качественное присутствие по характерному запаху. Более объективным качественным показателем служат свинцовые индикаторные бумажки (фильтровальная бумага, пропитанная раствором ацетата свинца). При опускании в воду, содержащую сероводород, свинцовая бумага темнеет, принимая желтую (малое содержание), бурую (среднее содержание) или темно-коричневую (высокое содержание) окраску.
В водных растворах сероводород присутствует в трех формах: недиссоциированный H2S, в виде ионов HS – и S 2 – . Относительные концентрации этих форм в воде зависят от рН этой воды и в меньшей степени от температуры и общего солесодержания.
Если анализируемая вода не содержит веществ, реагирующих с иодом, то сероводород и его ионы можно определить следующим образом.
В основе количественного метода определения H2S лежит реакция окисления сероводорода йодом:
К точно отмеренному подкисленному раствору йода, взятого в избытке по отношению к ожидаемому содержанию сероводорода, прибавляют определенное количество воды. Количество йода, израсходованное на окисление сероводорода, определяется обратным титрованием остатка йода тиосульфатом. Разница между количеством раствора тиосульфата, соответствующим всему количеству взятого для анализа йода, и количеством этого же раствора, затраченного на титрование остатка йода в пробе, эквивалентна содержанию сероводорода в исследуемой пробе.
источник